Published: July 2014
Last Reviewed: August 2017
There are many inherited metabolic diseases that may have a pathologic impact on the liver. In many cases, the liver component of these diseases is only an epiphenomenon of a more generalized systemic disorder. Examples of such epiphenomena are glycogen and lipid storage diseases, in which hepatomegaly is a manifestation of the underlying metabolic defect, although the liver is not necessarily the major target organ. However, there are three genetically determined diseases in which the liver may be the principal target organ, with manifestations of acute, subacute, or chronic disease that may become evident in early or later life. These are hereditary hemochromatosis (HH), a major disorder of iron overload, Wilson's disease, a genetic disorder of copper overload, and alpha1-antitrypsin (α1-AT) deficiency, a disorder in which the normal processing of a liver-produced protein is disturbed within the liver cell.
In some cases, the awareness of these conditions is brought about by suspicion based on a specific clinical syndrome. In other cases, these conditions have to be excluded when faced with nonspecific liver disease abnormalities, such as elevated liver enzyme levels, hepatomegaly, or previously undiagnosed portal hypertension. In the case of hemochromatosis, the approach to early diagnosis has moved one step further, with an awareness that markers of iron overload may be present in the serum long before liver disease has developed. These chapters will focus on discussions of these three conditions.
Certain key concepts (Box 1) are common to all three conditions and need to be emphasized at the outset. First, although the recognition of inherited liver disease is often the process of exclusion of more common causes (e.g., viruses, alcohol, autoimmunity), it is important to emphasize that awareness of the clinical features of these metabolic liver diseases should promote a proactive diagnostic evaluation. Second, inherited metabolic liver disease may manifest in childhood or may be delayed until adult life and, in some cases, may regress after the childhood or adolescent years, only to reappear later in life. Third, with the advent of molecular diagnostic testing, phenotypic assessment of these conditions may be now complemented in certain cases by genotypic evaluation. Fourth, with the availability of effective treatments, there has been a dramatic impact on the prognosis of metabolic liver diseases in both childhood and adult life, further emphasizing the importance of early diagnosis. Finally, in several conditions (e.g., α1-AT deficiency, Wilson's disease), liver transplantation corrects the primary biochemical abnormality in the liver and effectively cures the disease.
| Box 1: Key Concepts |
|---|
|
Hereditary hemochromatosis (HH) is defined as an inherited disorder of iron metabolism that leads to progressive, parenchymal, cellular iron overload in many tissues of the body–in particular in the liver, pancreas, and heart (Box 3). When the degree of iron overloading reaches a critical level, structural and functional damage to these organs may become apparent, and these constitute the phenotypic evidence for HH. The genotypic definition is based on a single missense mutation, the so-called C282Y mutation, of the HFE gene on the short arm of chromosome 6, which has a major role in the regulation of iron metabolism. When this mutation is present in both copies of the gene, homozygous HH is said to be present. Not every individual who is homozygous for this mutation develops phenotypic evidence of iron overload. Furthermore, there are other hereditary forms of iron overload based on alternative mutations of the HFE gene or mutations of other genes that may also play a role in the regulation of iron metabolism.
The HFE gene and its mutation were first described in 1996.The HFE gene has considerable homology with other major histocompatibility complex class 1 antigens. The principal mutation at amino acid position 282 in the protein product of this gene leads to the substitution of tyrosine for cysteine, which has a profound effect on the function of this protein. Retrospective analyses of individuals in the United States with phenotypic or familial evidence for homozygous HH have revealed that 83% to 100% were homozygous for the C282Y mutation. Another mutation, H63D, located at the 63 position of the HFE protein, was present in association with the C282Y mutation in approximately 4% of similar populations. These persons are termed compound heterozygotes. Subsequent studies from other countries have confirmed an average prevalence of C282Y homozygosity of approximately 90% in previously diagnosed HH patients. Approximately 10% of individuals with a clinical condition that is phenotypically similar to HH lack the C282Y mutation. Mutations of other genes not located on chromosome 6 also may play a role in iron metabolism and are currently being investigated.
| Box 3: Causes of Iron Overload |
|---|
|
Hereditary Hemochromatosis
Secondary Iron Overload
|
It is now evident that HH is the most common identified mendelian genetic disorder in the white population. Although its geographic distribution is worldwide, it is particularly common in individuals of Northern European descent, particularly of Nordic or Celtic ancestry. Overall, its prevalence in the white population is approximately 1 in 300, with a higher prevalence of 1 in 150 to 200 in the Anglo-Celtic-Nordic population.
Before mutational analysis was available, it was believed that the homozygous genetic abnormality inevitably led to progressive iron overload. With the availability of genotypic analysis, it is now apparent that the homozygous state for the mutation does not invariably lead to iron overload. There are those who have the genetic mutation on both alleles but who do not express it phenotypically. However, in those who do, there appears to be a pathophysiologic predisposition to increased inappropriate absorption of dietary iron, which leads to the progressive development of life-threatening complications of cirrhosis, hepatocellular cancer, diabetes, and heart disease. The normal HFE protein is expressed predominantly in the crypt cells of the upper intestine where, in association with the transferrin receptor, it may play a role in sensing the iron status of the body. Evidence has suggested that the mutant HFE protein is unable to provide this sensing, therefore “misinforming” the villous cell of the small intestine that iron deficiency might be present. This may lead to upregulation of the iron transport protein normally expressed on the villous cell, thereby explaining the inappropriate intestinal absorption of dietary iron. Although the genetic predisposition to increase iron absorption is present at birth, the disease may take 40 to 50 years or longer to progress to significant organ damage. Therefore, it is useful to think of the evolution of this clinical condition as a series of stages that begins with clinically insignificant iron accumulation based on the genetic abnormality (from 0 to 20 years of age, 0 to 5 g parenchymal iron storage). Subsequently, this evolves to a stage of iron overload without evident disease (at approximately 20 to 40 years of age, 5 to 20 g parenchymal iron storage). If left untreated, the condition may progress to the stage of iron overload with organ damage (usually at 40 years of age or older, and with more than 20 g of parenchymal iron storage).
With the increased awareness of hereditary hemochromatosis as a common clinical condition in whites, the disease is being diagnosed as a condition of predisposition to iron overload before the classic clinical symptoms and signs develop. In the past 40 years, the percentage of individuals with this condition who were symptomatic, with evidence of target organ damage, has been overtaken by a predominance of individuals who are asymptomatic, with only laboratory evidence for iron overload but with no abnormal physical signs. The classic triad of cirrhosis, diabetes mellitus, and skin pigmentation—so-called bronzed diabetes—is now a rare finding.
Even in the absence of early diagnoses based on abnormal serum iron markers, or on abnormal liver function found incidentally or by appropriate family screening, many early symptoms of HH are nonspecific. These consist of weakness, malaise, fatigue, lethargy, and weight loss that may not evoke an awareness, even in the astute clinician, unless appropriate laboratory tests are performed. At this stage, there may be no abnormal physical signs or only a minimal degree of hepatomegaly. Development of arthralgias, loss of libido, or impotence may also not arouse the suspicion of HH unless appropriate laboratory tests are performed. These features may antedate the more classic and specific clinical findings associated with involvement of the liver, pancreas, heart, and skin. At this later point in the development of the condition, usually when there is significant heavy iron overload, patients may reveal marked hepatomegaly, abnormal liver enzyme levels, skin pigmentation resulting both from iron deposition and increased melanin, glucose intolerance, and cardiac signs indicative of a dilated cardiomyopathy, with associated cardiac dysrhythmias and congestive heart failure.
As the liver disease progresses, portal hypertension with ascites, splenomegaly, and additional cutaneous features of chronic liver disease become apparent. These features of progressive liver disease are greatly accelerated in the face of coexisting risk factors such as alcohol abuse, hepatitis C, or nonalcoholic steatohepatitis. Features of hypogonadism may be difficult to interpret as specific for iron overload because they are common complications of end-stage liver disease.
The diagnostic approach to HH may be targeted at distinct populations (Box 4). These may be symptomatic individuals with unexplained features of liver disease, type 2 diabetes mellitus associated with hepatomegaly and elevated liver enzyme levels, and early-onset atypical arthropathy, cardiac disease, and male sexual dysfunction. In the asymptomatic group, one has to consider the diagnosis in the first-degree relatives of confirmed cases of HH in those with abnormal serum iron markers during routine testing, or other patients referred to the liver clinic with unexplained elevation of liver enzyme levels, asymptomatic hepatomegaly, or radiologic features on CT scanning or magnetic resonance imaging (MRI) suggestive of hepatic iron overload.
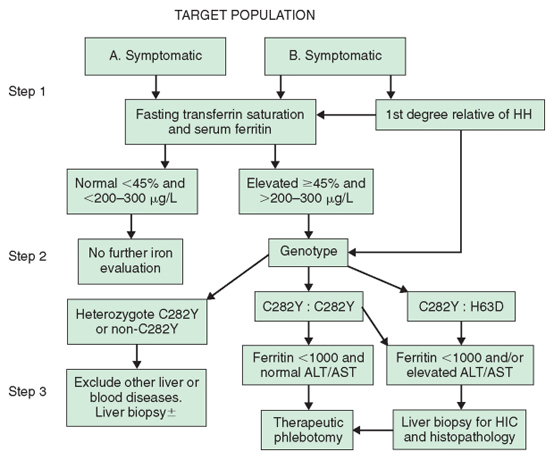
Because it is agreed that clinical HH is the result of iron overload, the diagnosis is based on the documentation of increased iron stores—namely, increased hepatic iron concentrations associated with elevated serum ferritin levels. As serologic iron markers have become more widely available in recent years, most patients with HH are now identified while still asymptomatic and without evidence of target organ damage. Now, with the availability of mutational analysis, HH may be further defined genotypically in a first-degree relative by the finding of C282Y homozygosity or C282Y-H63D compound heterozygosity. There is evidence to support the cost effectiveness of sequential testing of these target populations by a combination of indirect phenotypic markers of iron overload in the serum followed by mutational analysis to confirm the presence of classic HFE-related HH (Fig. 1). In this type of analysis, the cost effectiveness of such an approach remains valid, even assuming that as few as 20% of individuals with C282Y homozygosity will ever develop life-threatening complications of the disease.
| Box 4: Target Populations for the Diagnosis of Hereditary Hemochromatosis (HH) |
|---|
|
Data on the sensitivity, specificity, and positive and negative predictive value of phenotypic screening tests have been provided by studies of healthy blood donors, large-scale screening of a healthy population, and first-degree relatives of detected homozygotes. The diagnostic algorithm (see Fig. 1) from the guidelines adopted by the liver and gastroenterology subspecialty societies proceeds in three steps, beginning with phenotypic evaluation and followed by mutational analysis of those individuals with confirmed elevated serum iron markers.
The initial step in the diagnostic approach to HH is the fasting transferrin iron saturation (TS). This is the ratio of serum iron to total iron binding capacity, expressed in μg/dL multiplied by 100. An elevated TS value is the earliest phenotypic manifestation of HH. This is the relatively small circulating iron compartment that appears to be very sensitive to increased dietary iron absorption. The presence of an associated elevated serum ferritin level is usually indicative of accumulating iron stores. Both TS and serum ferritin are susceptible to nonspecific elevation, particularly in the presence of inflammatory diseases and other causes of liver disease. Earlier studies, based largely on evaluation of first-degree relatives of HH patients, have suggested that the combination of these tests has a high degree of sensitivity, specificity, and positive predictive value. Subsequent studies in unselected populations have suggested much lower degrees of sensitivity for both these markers in asymptomatic C282Y homozygotes. However, these tests may now be automated and are relatively inexpensive, providing the most effective means for the early detection of iron overload. Furthermore, a normal serum ferritin level in combination with a TS value lower than 45% has a negative predictive value of 97% and exceeds the accuracy of any of the indirect tests used in isolation. The decision to use a value of 45% for TS is based on studies that suggest values in excess of this correctly identify at least 98% of C282Y homozygotes. However, it is recognized that values in excess of 45% may often include C282Y heterozygotes and others with relatively minor degrees of secondary iron overload (e.g., those with alcoholic liver disease, nonalcoholic steatohepatitis, and chronic hepatitis C, and those who have had surgical portacaval shunts). Finally, the serum ferritin level may have an additional prognostic value; individuals with a ferritin level higher than 1000 ng/mL have a greatly increased likelihood of developing hepatic fibrosis or cirrhosis.
The second stage in the algorithm is genetic mutation analysis for the C282Y and H63D mutations of the HFE gene. This step is reserved for individuals with a fasting TS value higher than 45% and an elevated serum ferritin level. The presence of HFE mutations can now be detected by polymerase chain reaction testing on samples of whole blood, which is available commercially. Individuals with serum indicators of iron overload who are found to be homozygotes for the C282Y mutation are candidates for phlebotomy therapy; in those with no risk factors for significant liver injury, therapeutic phlebotomy may be undertaken without need for a liver biopsy. These usually include those younger than 40 years, with no clinical evidence of liver disease (e.g., increased liver enzyme levels, hepatomegaly) whose serum ferritin level is lower than 1000 ng/mL. Finally, the current recommendation is to offer mutation analysis to first-degree relatives of known HH patients, regardless of the phenotypic markers of iron overload. In such individuals, the presence of homozygosity for the major mutation provides an indication for subsequent regular evaluation of transferrin saturation and serum ferritin levels. Adults who are determined to be homozygous for the major mutation may be offered guidance about the likelihood of the presence of the mutation in their children by appropriate mutation analysis in the spouse. The failure to detect either mutation in the spouse offers reassurance that the children can be only obligate heterozygotes.
The third step of the algorithm advances evaluation through therapeutic phlebotomy. However, the option remains to perform a liver biopsy when there is a strong suggestion of liver disease. Furthermore, liver biopsy is also recommended for compound heterozygotes with elevated TS and abnormal liver enzyme levels or clinical evidence of liver disease. Liver biopsy has a value in the documentation of the presence of cirrhosis or other possible causes of liver disease responsible for elevated liver enzyme levels or hepatomegaly, thereby contributing to evaluation of prognosis in a patient with HH. Finally, the liver is the most easily accessible tissue for accurately assessing the level of iron stores. The degree and cellular distribution of iron stores are assessed using Perls' Prussian blue test, which provides a qualitative assessment of iron stores based on stainable iron. Grade IV staining is defined as deposition of iron in all zones of the acinus. In addition, quantitative iron determination may be made on fresh-frozen or formalin-fixed tissue. A hepatic iron concentration (HIC) of more than 1800 μg/g dry weight (equal to approximately 32 μmol/g) is indicative of excess iron in the tissue. Because excessive iron deposition in homozygotes who develop iron overload is usually a lifelong progressive accumulation, the rate of iron accumulation itself may provide powerful evidence of homozygous HH. A rate in excess of 1.9 μmol/g/yr is found in most HH homozygotes. This rate is defined as the hepatic iron index, which used to be regarded as the gold standard for homozygous HH. It is now recognized that up to 15% of genotypic homozygotes may have a rate of iron accumulation lower than 1.9 μmol/g/yr; such a definition, based on the hepatic iron index, is no longer regarded as essential for diagnosis. In individuals with HH who are older than 20 years, the HIC is usually at least three times the upper limit of normal (approximately 5400 μg/g), although it is recognized that spuriously low levels of hepatic iron may occur for various reasons, including voluntary blood donation. The HIC is usually elevated to levels exceeding 14,000 μg/g dry weight in those who have already developed hepatic fibrosis or cirrhosis; they are usually older than 40 years. In certain cases, these manifestations of liver damage may occur at a younger age and at lower levels of hepatic iron. In the absence of cofactors such as alcohol or hepatitis, it is rare to see fibrosis or cirrhosis in those younger than 40 years.
It is therefore important to emphasize that the value of liver biopsy is not limited to determination of the HIC. Rather, the documentation of cirrhosis on liver biopsy also has a significant impact on the prognosis in terms of morbidity and mortality in HH patients. Individuals who are treated before the development of cirrhosis usually have a normal life expectancy.
The treatment of HH is simple and relatively safe. Therapeutic phlebotomy will effectively mobilize and remove iron stores and, when adhered to on a regular basis, will maintain them at normal levels (Table 2). Patients should be encouraged to adhere to a regimen of phlebotomy of one unit of blood once or twice weekly as tolerated initially. This will remove approximately 250 mg of iron for each unit of phlebotomy, depending on the starting hematocrit value. In situations in which total body iron stores exceed 20 to 30 g, this regimen of phlebotomy may take up to 2 to 3 years to complete. The aim is to reduce iron stores to a level just short of iron deficiency. The hematocrit value should be monitored before each phlebotomy and should be postponed if it falls by more than 20% of its starting value. It is reasonable to check the serum ferritin level after every 10 to 12 phlebotomies. The serum ferritin level may be expected to fall progressively with iron mobilization, and it can be confidently assumed that effective mobilization of the iron stores will be completed when the serum ferritin level falls below 50 ng/mL.
| Initial Treatment | Maintenance Treatment |
|---|---|
|
|
Subsequently, a maintenance schedule may be initiated, and it can be expected that a one-unit phlebotomy may be necessary every 2 to 3 months. The aim of maintenance therapy is to keep the serum ferritin level between 25 and 50 ng/mL, thereby avoiding overt iron deficiency. Currently, phlebotomy is a therapeutic procedure, with a coding recognized by the Centers for Medicare and Medicaid Services (formerly the Health Care Finance Administration) and third-party insurers. Although there is continued elevation of the TS level until late in the course of therapy, it is important to avoid pharmacologic doses of vitamin C, which may result in accelerated mobilization of iron. This might saturate the circulating transferrin and lead to potentially toxic complications, such as cardiac dysrhythmias and cardiomyopathy. Deferoxamine (Desferal) is an iron-chelating agent reserved for those with secondary iron overload caused by dyserythropoietic anemia. Finally, there are many specialists who prescribe erythropoietin, which is given systemically to promote red cell production in those who are unable to mount a bone marrow response to phlebotomy.
There have been several longitudinal studies that have provided powerful evidence that initiation of phlebotomy therapy before cirrhosis and/or diabetes develop will significantly reduce the morbidity and mortality of HH. These data have provided the impetus for the treatment of asymptomatic individuals with homozygous HH and markers of iron overload and of others with evidence of potentially toxic levels of storage iron. In symptomatic patients, treatment is also indicated to attenuate progressive organ damage. There are certain clinical symptoms and signs that may be improved by phlebotomy (e.g., fatigue, malaise, skin pigmentation, abdominal pain, level of insulin requirements in diabetics). Other clinical features may be less responsive to iron mobilization (e.g., arthropathy, hypogonadism, established cirrhosis). Primary liver cell cancer accounts for about 30% of all iron-related deaths in HH, with another 20% ascribed to complications of cirrhosis.
Therefore, the preemptive treatment of asymptomatic patients before these complications develop may play a major role in the reduction of mortality from HH. What is more debatable in light of the available methods for mutation analysis is whether everyone who is a C282Y homozygote will inevitably develop iron overload, with its potential complications. Because the definition of HH is still based initially on phenotypic manifestations, there are no convincing arguments to treat genetically predisposed individuals who have no indirect markers of iron overload. This has been used as one of the proposed arguments against any recommendation at present for widespread genetic screening for hereditary hemochromatosis. Nevertheless, evidence points to a majority of genetically predisposed individuals—that is, C282Y homozygotes—particularly males exhibiting elevated iron markers. We do not have accurate predictors of how many of these will go on to develop tissue iron overload, and most specialists currently favor prophylactic phlebotomy in these cases.
Because cirrhosis in its fully developed form is not reversible by iron removal, the potential remains for decompensated liver disease in these individuals and may provide an indication for considering OLT. Mortality rates in HH patients who have had transplants are less satisfactory than in those who have had transplants for other causes of liver disease. Because most post-transplantation deaths in these patients occur in the perioperative period as a result of cardiac or infection-related complications, it has been suggested that every endeavor should be made to diagnose HH at an early enough time point to permit adequate removal of excess iron stores before OLT. Again, it should be emphasized that iron removal treatment in HH before the development of cirrhosis or diabetes can help maintain normal life expectancy, providing a persuasive argument for preventive therapy.