Published: April 2017
Last Reviewed: December 2017
Hereditary angioedema (HAE) is an inherited condition characterized by recurrent episodes of nonpruritic, nonpitting, subcutaneous or submucosal swelling without the presence of urticarial lesions. Multiple areas of the body can be involved including hands, feet, intestinal wall, genitalia, face, tongue, or larynx.1 Swelling of the pharynx or larynx can be life–threatening due to asphyxia.
The prevalence of HAE is estimated to be approximately 1 case per 50,000.2 Hereditary angioedema arises from gene mutations that encode an important protease known as C1 inhibitor belonging to a serine superfamily of proteins. More than 100 mutations have been reported to affect the C1–inhibitor gene, including up to 25% de novo mutations.3
There are 2 main types of HAE that are inherited in an autosomal dominant manner. Type I HAE accounts for approximately 85% of cases and results from a quantitative deficiency of C1 inhibitor. Type II HAE is responsible for approximately 15% of cases and results from a dysfunctional C1 inhibitor protein.
More recently, a third form of HAE has been identified as HAE with normal C1 inhibitor. This condition, formerly known as type III HAE, occurs mostly in females in whom both quantitative and functional studies of C1 inhibitor are normal. This condition has been thought to be associated with increased estrogen levels, although it is not exclusively found among females. In some cases, HAE is associated with a gain of function mutation in factor XII, a protein involved in the coagulation cascade.4
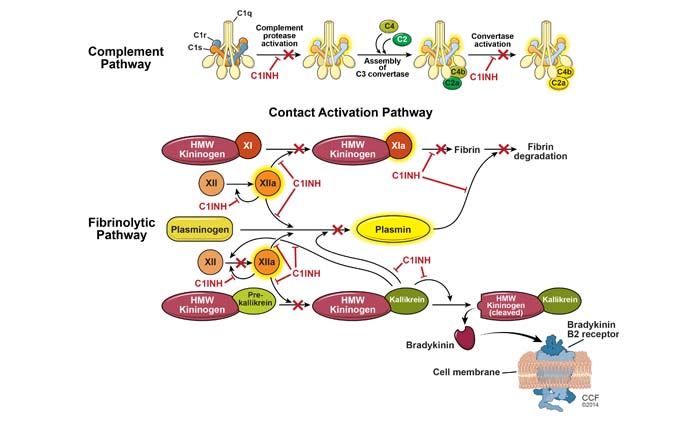
The principal mediator involved in HAE during episodes of swelling is bradykinin, a byproduct of the plasma contact system. The C1 inhibitor plays a significant role in the complement cascade, coagulation, and contact systems. As its name implies, it has an important inhibiting effect on the major proteases in these pathways, including activated Hageman factor, kallikrein, and plasmin, as illustrated in (Figure 1). The C1 inhibitor is ultimately responsible for regulating the production of bradykinin. In this manner, episodes of trauma or stress can activate contact and complement pathways. In the setting of C1–inhibitor deficiency (type I HAE) or C1–inhibitor dysfunction (type II HAE),5 increased levels of bradykinin lead to recurrent episodes of angioedema. HAE with normal C1 inhibitor has been associated with defects in the coagulation cascade, although its underlying pathophysiology remains to be determined.
The typical signs and symptoms of HAE start during childhood or puberty and persist throughout life. Usual attacks start with a prodromal sensation of tingling that can be accompanied by a nonpruritic wavy rash. This is followed by slowly progressive swelling that gradually subsides over 48 to 72 hours. The type of swelling seen in HAE is not associated with the presence of urticaria and it is not responsive to the use of steroids and/or antihistamines. Oropharyngeal swelling is of great concern as it can lead to asphyxia and death if it not recognized or treated appropriately. These type of attacks are less frequent, but more than half of HAE patients have at least 1 oropharyngeal attack during their life.6 Abdominal attacks carry significant morbidity and often require emergency visits, hospitalizations, and unnecessary procedures.
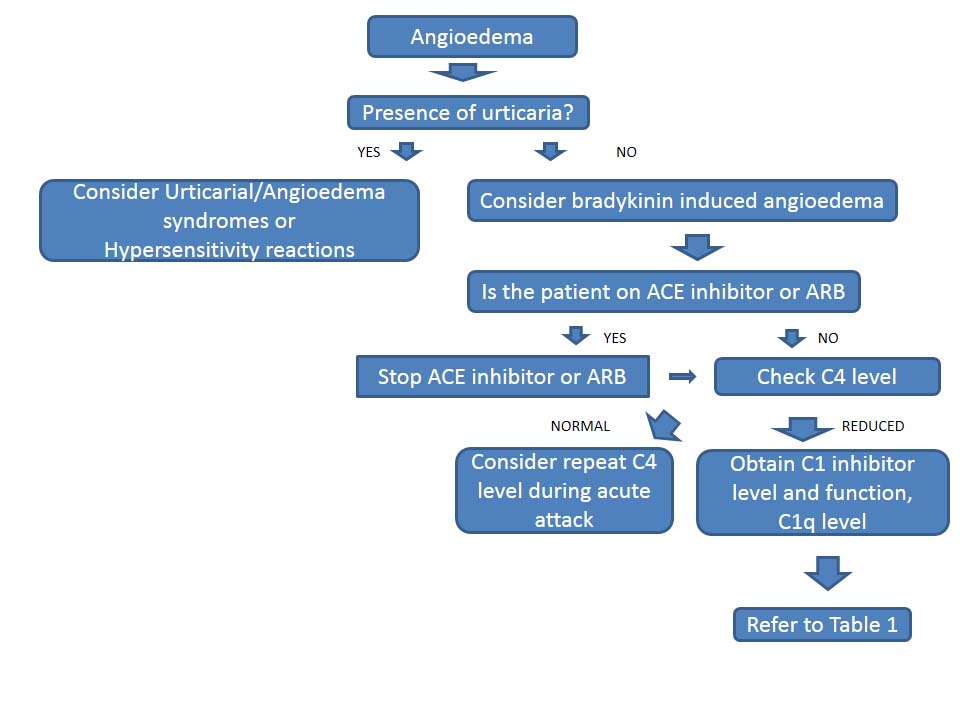
The diagnosis of HAE should be considered in individuals presenting with recurrent episodes of swelling, particularly if the swelling is not responsive to antihistamines or steroid therapy. The diagnosis should also be considered if swelling episodes are not associated with hives. Published guidelines for the diagnosis of HAE are available.7
The differential diagnosis of HAE includes the following:
Unlike HAE, the main mediator of angioedema in the setting of hypersensitivity reactions or urticaria/angioedema syndromes is histamine, not bradykinin. Bradykinin is responsible for recurrent episodes of swelling seen in HAE and in acquired angioedema and ACE inhibitor–induced angioedema syndromes.
Defects in C1 inhibitor lead to unregulated breakdown of the complement cascade including low levels of C4, C1 inhibitor, and/or C1 inhibitor function, seen at baseline and during an acute attack. Rarely, C4 levels remain normal at baseline but decrease in the setting of an acute attack. Acquired angioedema syndromes related to underlying proliferative disorders are often associated with low levels of C1q, as shown in (Table 1). In HAE with normal C1 inhibitor, levels are normal at baseline and during attacks. Gene mutation analysis of factor XII (Hageman factor) may establish the diagnosis; however, an unremarkable result does not exclude HAE with normal C1 inhibitor. There are no laboratory tests that can confirm HAE with normal C1 inhibitor or ACE inhibitor–induced angioedema (Figure 2).
| C4 | Antigenic C1 inhibitor | Functional C1 inhibitor | C1q | |
|---|---|---|---|---|
| Hereditary angioedema | ||||
| Type I | Low | Low | Low | Normal |
| Type II | Low | Normal–high | Low | Normal |
| With normal C1 inhibitor | Normal | Normal | Normal | Normal |
| Acquired angioedema | Low | Low–normal | Low–normal | Low |
| ACE inhibitor–induced angioedema | Normal | Normal | Normal | Normal |
ACE = angiotensin–converting enzyme. Adapted from Zuraw et.7
Treatment of HAE relies on long–term prophylaxis, short–term prophylaxis, and the treatment of acute attacks. The long–term goal is to minimize the frequency and severity of angioedema episodes. It has been reported that patients with HAE develop 1 to 2 swelling attacks per month on average.8
Long-term prophylaxis should be considered in patients with a history of life–threatening attacks or serious attacks more than once every 3 months. Prior to U.S. Food and Drug Administration (FDA) approval of C1 inhibitor replacement therapy in 2008, prophylactic or long–term treatment of HAE relied on the use of synthetic 17-α–alkylated androgens: danazol and stanozolol (Winstrol). In double–blind studies, androgens were associated with an increase in the level of C1 inhibitor and a reduction in the number of angioedema attacks.9,10 However, androgens are associated with risks related to their mechanism of action. Common side effects include masculinization, alopecia, acne, changes in lipid profiles, and risk for hepatic adenoma. For this reason, the use of synthetic androgens is not recommended for patients under the age of 16 years of age. Also, it is contraindicated for use in pregnant or breastfeeding women.11
Patients who are currently using anabolic androgens should have liver enzymes, urinalysis and lipid profile checked every 6 months and liver ultrasound evaluations every 6 to 12 months, depending on the dose. Dosing relies on finding the lowest dose required to reduce the frequency and severity of swelling episodes rather than targeting to a particular C4 or C1 inhibitor level. Danazol at 200 mg daily or every other day, or stanozolol at 2 mg daily or every other day, are considered effective doses for most patients.
Antifibrinolytic agents, such as aminocaproic acid, are another option for long–term treatment. Usual doses consist of 1 to 2 grams 3 times a day, although these drugs are not FDA approved for HAE treatment. Tranexamic acid has been used, but it also is not FDA approved for HAE treatment. Antifibrinolytic agents are reserved for patients who cannot tolerate anabolic steroids or other forms of prophylactic therapy.12,13 Side effects associated with these agents include myalgias, fatigue, and coagulation disorders.
More recently, various C1 inhibitor concentrates have been studied and FDA–approved for treatment of HAE in the United States. C1 inhibitor replacement therapy has been used successfully in Europe for several decades. Initial studies reported the effectiveness of C1 inhibitor replacement therapy.14,15 In 1989, data were published showing that use of purified C1 inhibitor increased the levels of C1 inhibitor, C2, C4, and CH50 and reduced the number and severity of attacks.16
In 2008, a human nanofiltered, pasteurized C1 inhibitor concentrate (Cinryze) was approved by the FDA for prophylactic treatment of HAE. The nanofiltration process adds a step to purify plasma–derived products in an effort to maximize protection against viral and, possibly, prion disorders such as Creutzfeldt–Jakob disease.17 A crossover trial in 22 patients with history of at least 2 HAE attacks per month showed that treatment with this nanofiltered C1 inhibitor concentrate significantly reduced the number of attacks over 12–week periods (6.26 vs 12.72 in the placebo arm; P<.001).18
This product is indicated for prophylactic use in adolescents and adults with HAE at a dose of 1000 units (10 mL) IV every 3 to 4 days. It is classified as category C for the treatment of HAE during pregnancy. It is also approved for self–administration at home. Hypersensitivity reactions and thrombotic events have been reported.
The FDA has not approved the use of human purified and nanofiltered C1 inhibitor concentrate for treatment of acute attacks although it has been shown to reduce the median time to onset of relief compared with placebo. In 2014, the cost of C1 inhibitor concentrate (Cinryze) was $4,906 per single dose in the U.S. According to the product prescribing information, the most common adverse reactions observed by at least 5% of subjects are upper respiratory tract infection, sinusitis, rash, and headache.
Angioedema attacks are most often triggered by trauma or stress. Therefore, patients with HAE require additional protection perioperatively and, in particular, prior to undergoing dental procedures in efforts to minimize the risk of oropharyngeal swelling. For this purpose, the use of C1 inhibitor replacement, high–dose anabolic androgens, and fresh frozen plasma has been successful.19,20 Common preoperative regimens include the following:
Fresh frozen plasma (FFP) contains C1 inhibitor and can be helpful to reduce severity of attacks. However, the use of FFP is considered controversial as it also contains proteases that can exacerbate symptoms during an acute attack.
Berinert is a human, pasteurized, plasma–derived C1 inhibitor product that was first FDA approved in 2009 for the treatment of acute facial, laryngeal, or abdominal attacks in adolescents and adults. It most recently gained FDA approval for pediatric patients in July 2016. This form of C1 inhibitor was found to shorten the median time–to–onset of relief from an acute attack from 1.5 hours to 0.5 hours (P=.0025 vs placebo) in a double–blind, placebo controlled study of 125 patients with HAE.21 Dosing is 20 U/kg IV. Symptom relief is usually seen within 30 to 60 minutes. This form of C1 inhibitor concentrate is not approved for long–term prophylaxis. There are reports of hypersensitivity reactions, laryngeal edema, and thromboembolic events with the use of this agent. However, the most commonly reported adverse effect compared with placebo was dysgeusia.
Plasma kallikrein is activated by protease coagulation factor XII. These factors are both inhibited by C1 inhibitor. Active plasma kallikrein induces production of bradykinin. In 2009, ecallantide (Kalbitor), a kallikrein–inhibitor synthesized in yeast, was approved to treatment acute HAE attacks in patients 12 years and older. Double–blind, placebo–controlled trials have shown statistically significant improvements at 4 hours for active treatment compared with placebo22,23 Ecallantide dosing is 30 mg (3 mL) subcutaneously in the setting of an acute attack. There are reports of anaphylaxis related to the development of anti–ecallantide antibodies after repeated exposure. Therefore, ecallantide must be administered in a healthcare setting where personnel, equipment, and supplies for treatment of anaphylaxis are present.
Icatibant (Firazyr), a selective bradykinin type 2–receptor antagonist, was FDA–approved in 2011 for the treatment of acute attacks in patients 18 years and older. Dosing is 30 mg (3 mL) administered subcutaneously in a single injection.
The first trials found statistically significant decreased time to symptom relief with the use of icatibant compared with tranexamic acid, but not compared with placebo. Subsequently, pooled data demonstrated that icatibant offered symptom relief to a greater percentage of subjects within 4 hours of use as well as provided quicker relief of symptoms compared with placebo and tranexamic acid.23,24 In 2011, a third trial found that icatibant reduced times of symptom severity and shortened the time to initial symptom relief of cutaneous or abdominal attacks (0.8 vs 3.5 hours; P<.001).25 Icatibant was approved for self–administration, although laryngeal attacks can occur. The most common side effects included localized reactions at the administration site.
The newest treatment option is a recombinant C1 inhibitor (Ruconest) that was FDA–approved in July 2014 to treat acute attacks of HAE in adolescents and adult patients. It is also category B in pregnancy (ie, studies have failed to demonstrate a risk to the fetus and there are no adequate and well–controlled studies in pregnant women). This drug is purified from milk of genetically modified rabbits, and, thus, it is contraindicated for patients with known or suspected hypersensitivity reactions to rabbits. The product is intended for self–administration at 50 IU per kg for patients weighing less than 84 kg and 4200 IU for patients weighting greater than 84 kg. Approval was based on randomized, placebo–controlled trials and open–label extension showing significantly shortened time to initial relief (90 vs 152 mins; P=.031) based on treatment questionnaires and (75 vs 303 mins; P=.003) based on severity visual analog scale scores.26
Practice guidelines, including those for children, adolescents and women, have been published.7,27-29 (Table 2) summarizes the therapeutic options for HAE prophylaxis. (Table 3) summarizes treatment of acute attacks.
| Treatment | Dosing | Main side effects | FDA-approved Adults Children |
Pregnancy designation | Long-term prophylaxis | Short-term prophylaxis | Acute attacks | |
|---|---|---|---|---|---|---|---|---|
| 17 α alkylated androgens | ||||||||
| Danazol (Danocrine) | 200 mg/day or every other day | Masculization Alopecia Acne Hepatic adenomas Lipid abnormalities |
Yes | No |
Category X | Yes | Yes | No |
| Stanozolol (Winstrol) | Usual doses
Adults: 2 mg/day
Children 6–12 yr: 0.5–2 mg/day Children <6 yr: 0.5–1 mg/day |
Yes | Yes |
Yes | Yes | No | ||
| Antifibrinolytics | ||||||||
| Epsilon aminocaprioic acid (EACA) or Amicar | 1–2 g by mouth 3x daily | Hypercoagulability Muscle cramps Postural hypotension |
No | No |
Category C | Off-label use | ||
| Tranexamic acid (Cyklokapron) | 1g–.5g by mouth 2–3x daily | No | No |
Category B | Off-label use | |||
| Plasma derivatives | ||||||||
| Fresh frozen plasma | 2 U intravenous prior to procedure or during attack | Possible worsening of angioedema during an attack | No | No |
Off-label use | Off-label use | ||
| C1 inhibitor concentrates | ||||||||
| Plasma-derived nanofiltered C1 inhibitor (Cinryze) |
1000 U intravenous every 3–4 days | Hypersensitivity Possible transmission of infectious or prion disease Upper respiratory infection Rash Headache |
Yes* | Adolescents | Category C | Yes | Yes | No |
*self-administration
Adapted from Zuraw.1
| Treatment | Dosing | Main side effects | FDA-approved Adults Children |
Pregnancy designation | Long-term prophylaxis | Short-term prophylaxis | Acute attacks | |
|---|---|---|---|---|---|---|---|---|
| Plasma-derived purified C1 inhibitor (Berinert) | 20 U/kg intravenous on demand | Hypersensitivity reactions Possible transmission of infectious or prion disease Laryngeal edema Thromboembolic events Dysgeusia |
Yes* | All age groups |
Category C | No | No | Yes |
| Recombinant C1 inhibitor (Rocunest) | 50 IU/kg (<84 kg) 4200 IU (>84 kg) |
Hypersensitivity Headache Nausea Diarrhea |
Yes* | Adolescents | Category B | No | No | Yes |
| Plasma kallikrein inhibitor | ||||||||
| Ecallantide (Kalbitor) | 30 mg subcutaneous on demand | Anaphylaxis | Yes | 12 years and older | Category C | No | No | Yes |
| Selective bradykinin receptor antagonist | ||||||||
| Icatibant (Firazyr) | 30 mg subcutaneous on demand | Laryngeal attacks Localized reaction | Yes* | No | Category C | No | No | Yes |
*self-administration
Adapted from Cicardi et al.30
An unacceptably high mortality rate has been observed in patients with untreated or undertreated HAE. A 2012 study identified 214 deaths among 728 patients in families with C1 inhibitor deficiency.31 The same study estimated that approximately one–third of the deaths were from asphyxiation (N = 70) and 63 of those subjects died before HAE was diagnosed. With proper therapy, the morbidity and mortality of hereditary angioedema can be significantly reduced.
Treatment of HAE poses a special challenge in pregnant women and children. Attenuated androgens are contraindicated in both of these groups. Stanazolol, a 17 α–alkylated androgen, was the only FDA–approved agent for the long–term treatment of HAE in children, but it is no longer available in the U.S. In addition, its use poses risks common to all androgens, including the risk of premature puberty. Unfortunately, the most recent clinical trials done using other forms of treatment did not include the participation of children or pregnant women. Therefore, the newly FDA–approved therapies are mostly for the use of adolescents and adults.32
Therapy for the management of attacks in children with HAE must be individualized comparing the potential benefits versus the potential harms, and the burden of therapy. Fortunately, prepubertal children have been reported to have lower frequency and severity of attacks, and a "wait and see" or symptomatic treatment approach may be a reasonable option.28
There may be an increased frequency of HAE attacks in pregnant women, particularly during the second and third trimesters. Such attacks have been treated effectively with the use of C1 inhibitor concentrate.33 More recently, recombinant C1 inhibitor replacement with Rocunest has become an option for pregnant women, as it is classified as category B pregnancy in the United States. Risks, benefits, and burdens merit consideration in determining the most appropriate therapy.
Hereditary angioedema is an autosomal dominant disorder associated with substantial morbidity and mortality if untreated or undertreated. Deficiencies in either quantitative or functional levels of C1 inhibitor lead to increased production of bradykinin, the main mediator of recurrent episodes of swelling. New modes of treatment that specifically target affected pathways and lower frequency and severity of attacks have recently received FDA approval.
Roxana Siles, MD; nothing to disclose.