Published: August 2010
Last reviewed: June 2017
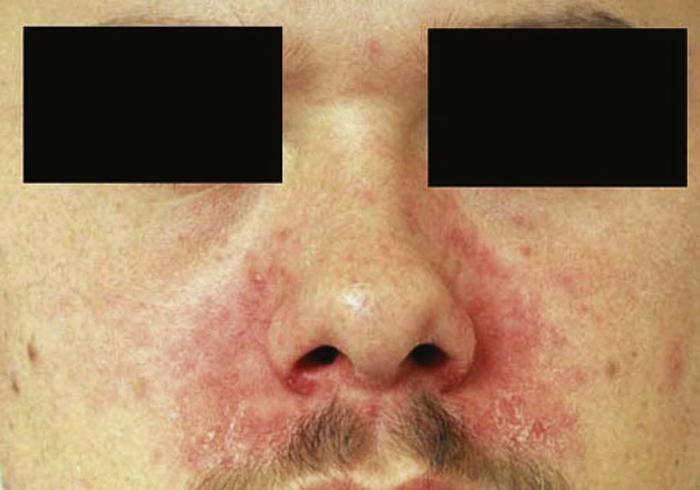
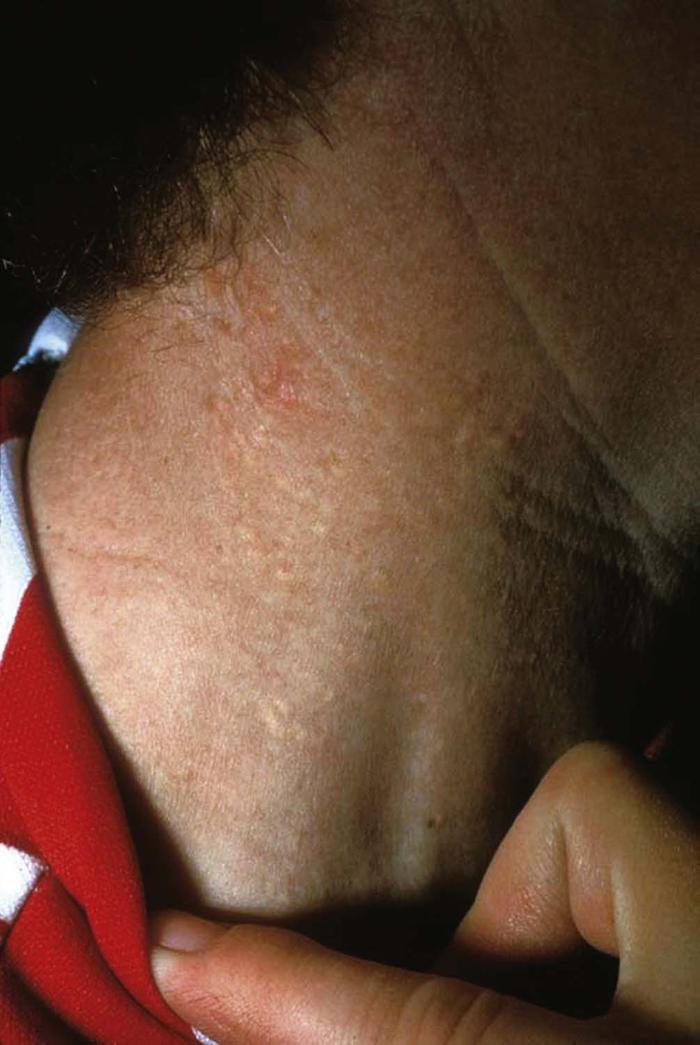
Seborrheic dermatitis (Fig. 1) is a common chronic, superficial inflammatory disease of the scalp, face (especially the eyebrows and nasolabial folds), ears, and central chest, affecting 2% to 5% of the population. Clinically, the disease is characterized by thin erythematous plaques, often with a fine, greasy scale. Pruritus is common and can be severe.
Recognition of seborrheic dermatitis is important for the primary care physician, because it may be associated with systemic disease, such as Parkinson's disease and human immunodeficiency virus (HIV) infection. Patients who have had a cerebrovascular accident (CVA) can develop seborrheic dermatitis on the scalp in a unilateral distribution, corresponding to the affected hemisphere. The pathophysiology of this phenomenon is not completely understood.
Differential diagnosis includes psoriasis, atopic dermatitis, allergic or irritant contact dermatitis, and dermatophyte (tinea) infections.
Treatment includes medicated shampoos containing zinc pyrithione, selenium sulfide, salicylic acid, coal tar, or ketoconazole in combination with topical corticosteroids. Alternatively, fluconazole 400 mg (one dose) may be effective in combination with a mild topical corticosteroid.
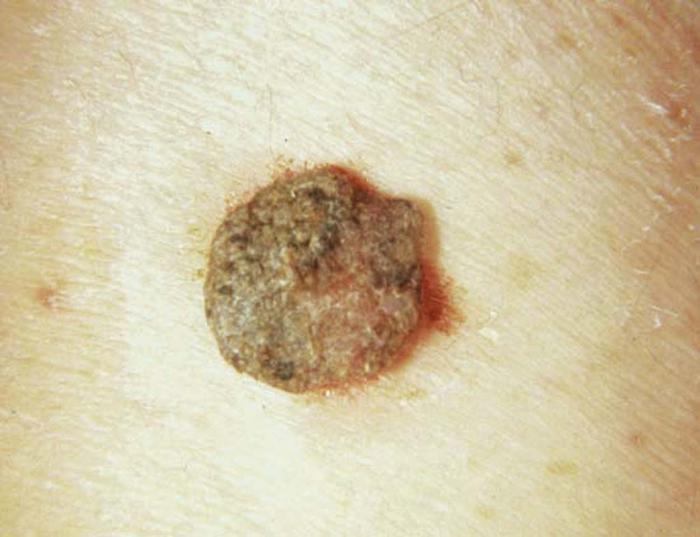
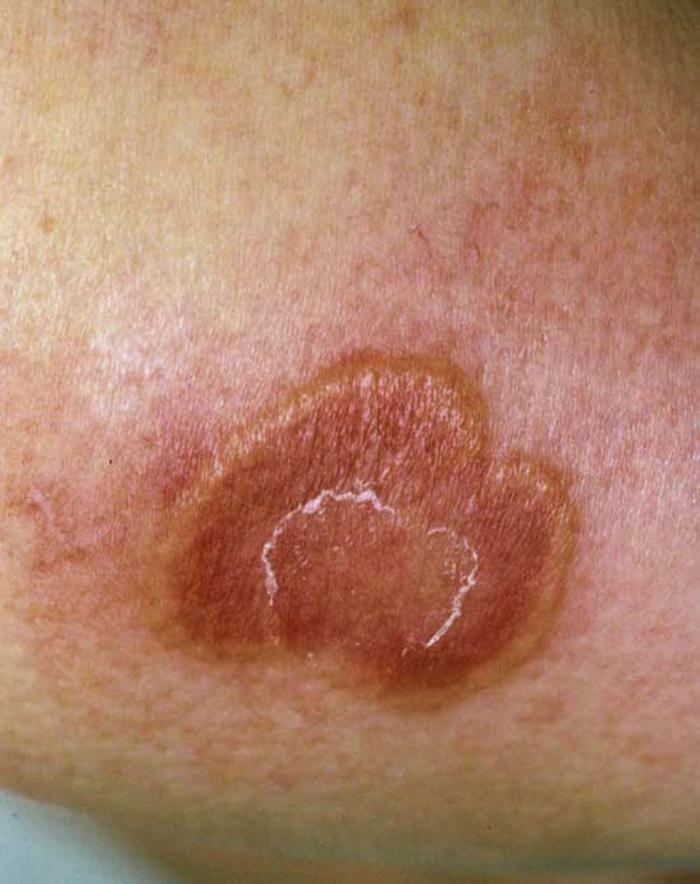
Seborrheic keratoses (Fig. 2), the most common benign cutaneous neoplasms, are warty, age-related hyperkeratotic papules and plaques that appear anywhere on the body, most commonly the trunk. Rarely, seborrheic keratoses indicate an underlying adenocarcinoma of the gastrointestinal tract if they appear suddenly in great numbers (sign of Leser-Trélat).
Differential diagnosis includes verruca vulgaris (warts), epidermal nevus, melanocytic nevi, and melanoma.
No treatment is necessary. If the plaques are pruritic, they can removed by curettage or cryotherapy.
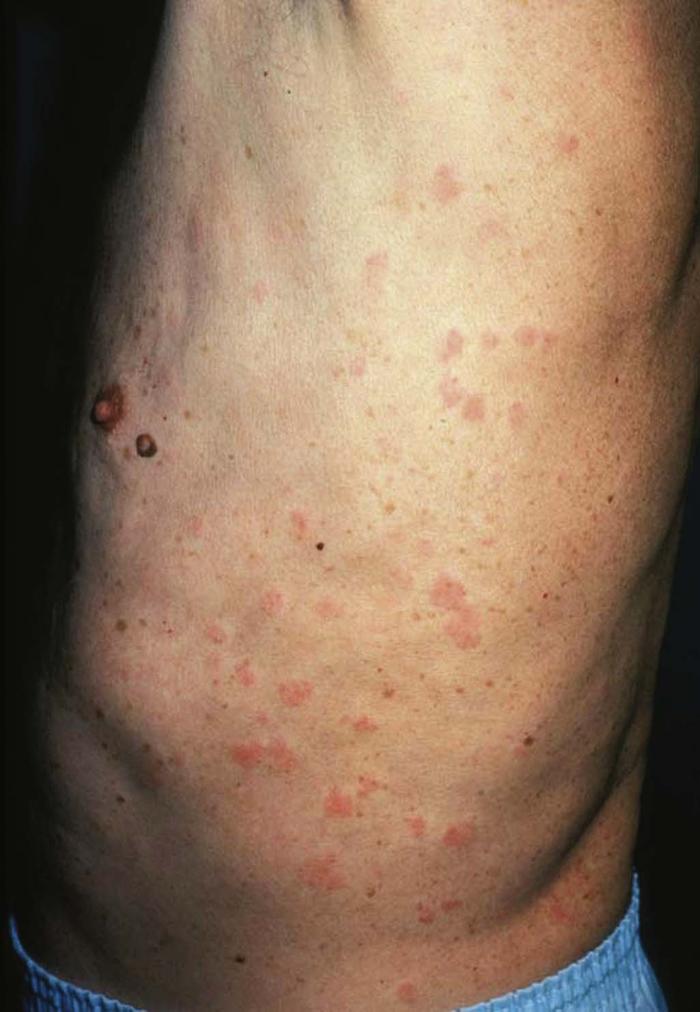
Urticaria (Fig. 3), or hives, is most often caused by medication (commonly penicillin or other antibiotics, sulfa drugs, aspirin) or food (shellfish, nuts, chocolate), and less often by infection. Hives are pruritic, edematous, evanescent wheals that resolve within 24 hours.
Acute urticaria typically lasts less than 6 weeks. Wheals in a fixed location for more than 24 hours suggest the possibility of urticarial vasculitis and warrant a skin biopsy. Chronic idiopathic urticaria for which no trigger can be identified often requires further testing such as serum radioallergosorbent testing (RAST) or skin prick-patch testing.
Differential diagnosis includes erythema multiforme, systemic lupus erythematosus (SLE), bullous pemphigoid, mastocytosis.
Treatment includes elimination of known causes, antihistamines (H1 and H2 blockers), oral corticosteroids for acute flares, and, in refractory cases, immunosuppresants such as sulfasalazine and cyclosporine.
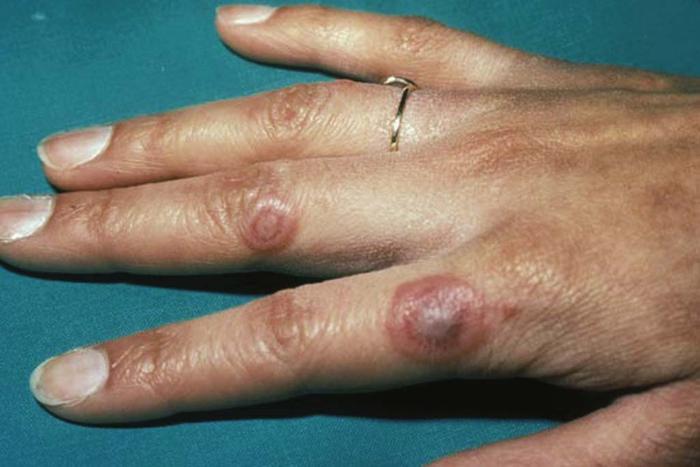
Erythema multiforme (Fig. 4), a cutaneous hypersensitivity reaction, is usually caused by infection (herpes simplex virus or Mycoplasma pneumoniae) and less commonly by drug sensitivity (sulfonamides, barbiturates, antibiotics). Division of erythema multiforme into two subsets based on clinical severity has been proposed: erythema multiforme minor and erythema multiforme major or Stevens-Johnson syndrome.1 Macules, papules, plaques, vesicles, or bullae, often with a targetoid or iris appearance, occur on the skin, often with an acral distribution (extremities). Erythema multiforme can also occur on mucosal surfaces. Prodromal symptoms are uncommon. Erythema multiforme minor is self limited and usually resolves within 2 to 4 weeks.1
Differential diagnosis includes urticaria, bullous arthropod reaction, drug eruption, and bullous pemphigoid.
Treatment includes suppressive oral antiviral agents such as acyclovir or valacyclovir, for herpes infection; discontinuation of possible causative medications; and supportive care.
Vitiligo (Fig. 5) is characterized by a focal or generalized distribution of depigmented macules and patches. The hairs in the vitiliginous areas are usually white. Vitiligo commonly occurs in periorificial areas (mouth, orbits, vagina, anus) or at sites of trauma (hands, elbows, knees). The disorder is often associated with autoimmune thyroid disease, insulin-dependent diabetes mellitus, pernicious anemia, or Addison's disease.
Differential diagnosis includes tinea versicolor, pityriasis alba, postinflammatory hypopigmentation, and hypopigmented mycosis fungoides.
Treatment includes broad-spectrum sunscreens, potent topical corticosteroids, topical calcineurin inhibitors (tacrolimus or pimecrolimus), narrow band ultraviolet (UV) B phototherapy, psoralen with UVA (PUVA) therapy, or total depigmentation for extensive disease.
Erythema nodosum (Fig. 6), the most common type of panniculitis, is characterized by painful, erythematous nodules on the shins and occasionally elsewhere. Erythema nodosum occurs most commonly in young women, with a peak incidence between 20 and 40 years.1 In addition to the cutaneous findings, patients can have fever, malaise, arthralgias, or arthritis. Typically the eruption is self limited, lasting an average of 3 to 6 weeks.1
The most common cause of erythema nodosum in the pediatric population is streptococcal pharyngitis. Other infectious causes include tuberculosis, gastrointestinal (GI) infections with Yersinia, Salmonella, or Shigella, and systemic fungal infections. Less common causes include drug sensitivity (sulfonamides, salicylates, iodides, oral contraceptives or hormone replacement therapy), and a variety of systemic diseases, most often inflammatory bowel disease (Crohn's disease more than ulcerative colitis) and sarcoidosis.
Differential diagnosis includes nodular vasculitis and other types of panniculitis.
Treatment includes identifying and eliminating known causes, bed rest and elevation of the extremities, aspirin or nonsteroidal anti-inflammatory medications (NSAIDs), colchicine, and supersaturated potassium iodide.
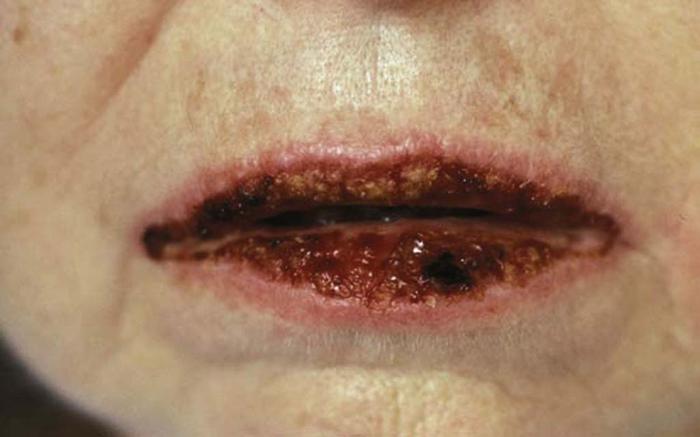
Pemphigus vulgaris (Fig. 7) is an uncommon chronic and debilitating blistering disease characterized by painful mucosal erosions and flaccid blisters that become erosive. Ninety percent of patients have mucosal disease, and erosions can outnumber intact bullae. Biopsy reveals characteristic suprabasilar acantholysis and intraepidermal bullae formation. Direct immunofluorescence reveals a chicken-wire pattern of deposition of immunoglobulin (Ig) G within the epidermis.
Pemphigus vulgaris can develop at any age, but it most commonly occurs in the fourth to sixth decades of life, usually in people of Mediterranean or Jewish ancestry.2 Morbidity and mortality are significant, even with treatment.
Differential diagnosis includes bullous pemphigoid, Stevens-Johnson syndrome, and epidermolysis bullosa acquisita.
Treatment includes good wound care for affected skin, systemic corticosteroids, various steroid-sparing immunosuppressants, rituximab, intravenous immunoglobulin (IVIg), and plasmapheresis.
Bullous pemphigoid (Fig. 8) is the most common bullous disease and is characterized by large, tense subepidermal blisters, which are often pruritic. Mucosal disease is rare. Biopsy reveals subepidermal bullae and an infiltrate of eosinophils. Direct immunofluorescence reveals a linear deposition of IgG at the dermal-epidermal junction.
Bullous pemphigoid occurs most commonly in the elderly, with an onset between 65 and 75 years of age. Prognosis is influenced by age and general condition of the patient, not by extent of disease activity.3 Treatment of older patients in poor health requires caution.
Differential diagnosis includes bullous SLE, epidermolysis bullosa acquisita, cicatricial pemphigoid, and dermatitis herpetiformis.
Treatment includes topical and systemic corticosteroids, steroid-sparing immunosuppressants, and tetracycline in combination with niacinamide.
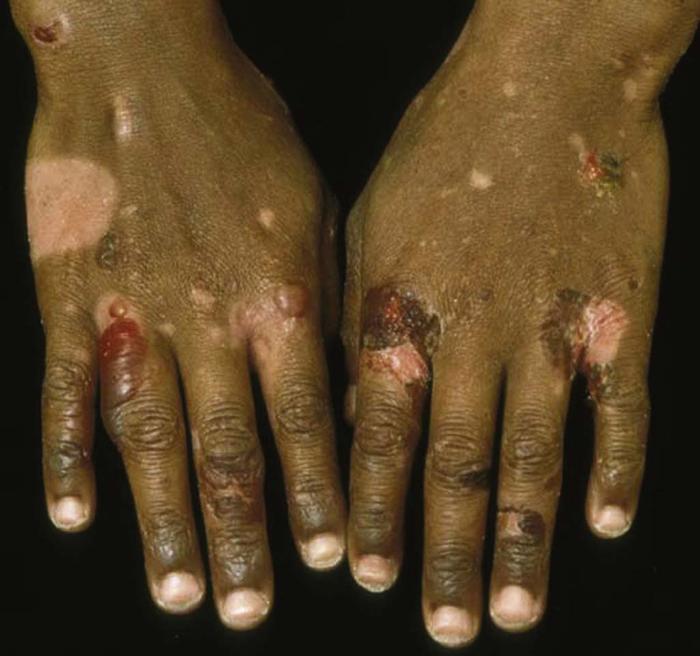
Epidermolysis bullosa acquisita (Fig. 9) is an uncommon bullous disease characterized by skin fragility, milia (small cysts), scarring alopecia, and nail dystrophy. Skin disease typically follows trauma and occurs primarily on the hands, feet, elbows, and knees. Immunofluorescence is similar to bullous pemphigoid, with IgG deposition at the dermal-epidermal junction.
Differential diagnosis includes bullous pemphigoid and bullous SLE.
Treatment includes topical and systemic corticosteroids, steroid-sparing immunosuppressants, colchicine, and plasmapheresis.
Cutaneous metastases (Fig. 10), are uncommon, and the reported prevalence varies from 0.7% to 10% of all patients with cancer.4 Any malignant neoplasm can metastasize to the skin. Cutaneous metastases from cancers of the lung, large intestine, and kidney are most commonly found in men; cancers of the breast and large intestine are the most likely primary tumors to metastasize to the skin in women.4 Metastases to the skin are usually flesh-colored to violaceous nodules that appear in close proximity to the primary neoplasm; most common sites are the head (scalp), neck, and trunk.
Differential diagnosis includes pilar or epidermal inclusion cyst, adnexal tumor, neurofibroma, and lipoma.
Treatment depends on the primary neoplasm and overall prognosis.
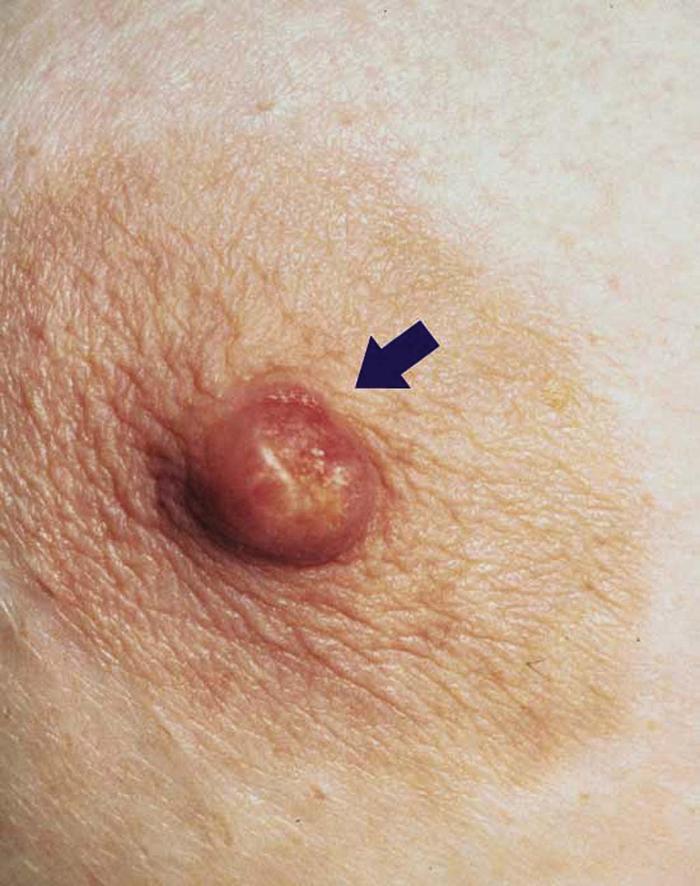
Paget's disease of the breast (Fig. 11) is an uncommon condition characterized by unilateral eczematous plaque of the nipple and areola. The disease is strongly associated with an underlying invasive carcinoma of the affected breast or ductal carcinoma in situ (DCIS).5
Extramammary Paget's disease is typically a persistent, eczematous plaque of the anogenital or axillary regions whose morphology and histology strongly resemble Paget's disease of the breast. Extramammary Paget's disease affects older adults and is often associated with an underlying adnexal (apocrine) carcinoma or an underlying cancer of the genitourinary tract or distal gastrointestinal tract.
Differential diagnosis includes allergic or irritant contact dermatitis (especially if bilateral), psoriasis, and dermatophyte (tinea) infection.
Treatment includes surgical excision, radiation therapy, and photodynamic therapy. Referral to oncology is recommended.
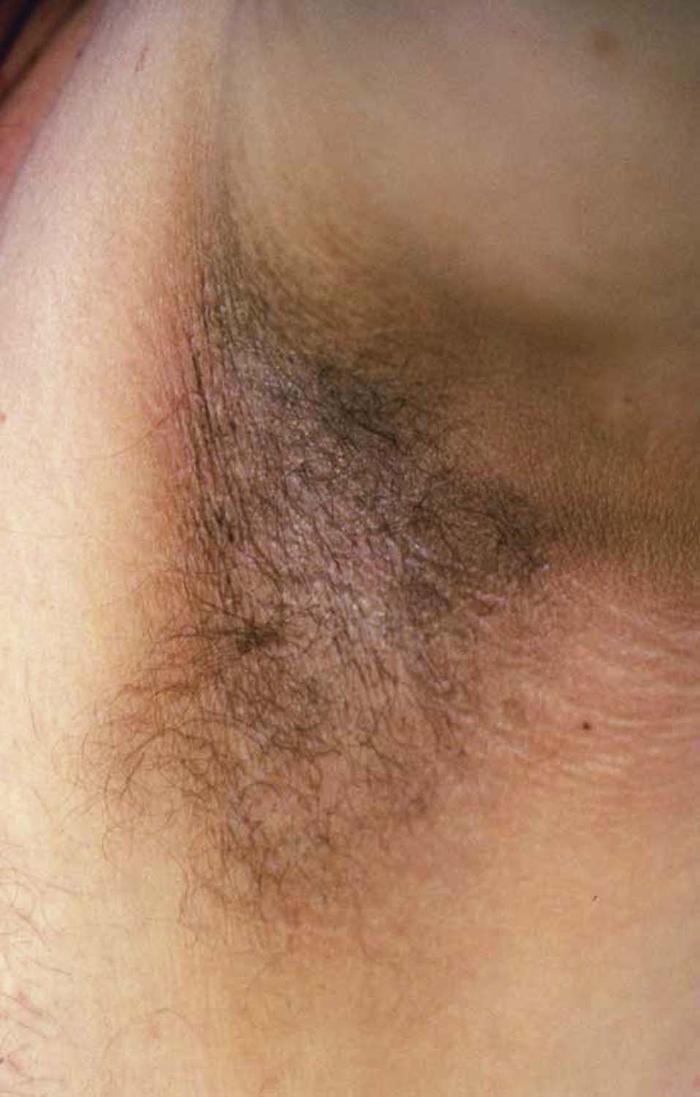
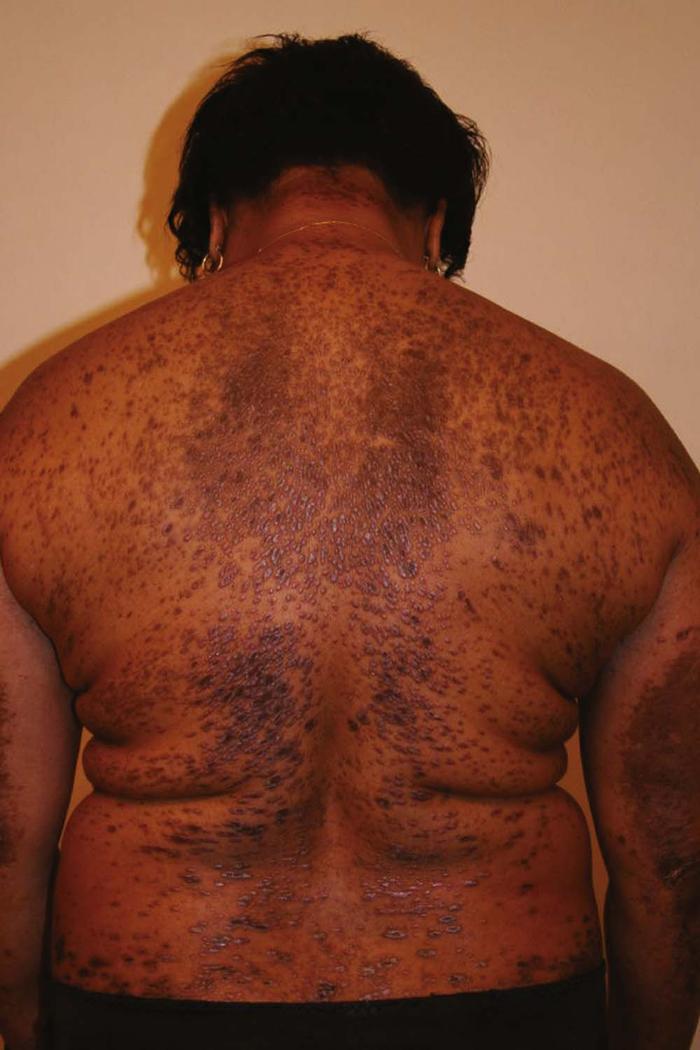
Acanthosis nigricans (Fig. 12) is characterized by smooth, velvet-like, hyperkeratotic plaques in intertriginous areas (e.g., groin, axillae, neck). Three types of acanthosis nigricans have been recognized.
Type I is associated with malignancy. Occasionally, acanthosis nigricans is a marker of an underlying adenocarcinoma, especially of the gastrointestinal tract (60% gastric). Malignant acanthosis nigricans has a sudden onset and more extensive distribution, including the face, palms, and trunk. Type II is the familial type, with autosomal dominant transmission. It is very rare and appears at birth or soon after. Type II has no malignancy association. Type III acanthosis nigricans is associated with obesity and insulin resistance. Type III is the most commonly occurring type.
Acanthosis nigricans can develop following the use of some medications, such as systemic corticosteroids, nicotinic acid, diethylstilbestrol, and isoniazid (INH).
Differential diagnosis includes confluent and reticulated papillomatosis of Gougerot and Carteaud and Dowling-Degos disease.
Treatment for type I acanthosis nigricans includes identifying and removing the malignant tumor. Treatment for types II and III includes weight loss and treatment of the underlying endocrine disorder, if applicable. Topical treatments including tretinoin, calcipotriol, urea, and salicylic acid may be helpful.
Cowden's syndrome, an autosomal dominant cancer syndrome caused by mutations in the tumor suppressor gene PTEN, is characterized by multiple tricholemmomas (wartlike growths) around the mouth, nose, and ears; cancer of the breast, endometrium, or thyroid gland; and thyroid disease (adenomas or goiter), mental retardation, and fibrocystic disease of the breast.
Sweet's syndrome (Fig. 13), or acute febrile neutrophilic dermatosis, has a strong association with acute myelocytic or myelomonocytic leukemia. Affected patients, usually middle-aged women, have painful erythematous to violaceous plaques on the face, extremities, and trunk. Most have fever, malaise, arthralgias, myalgias, and conjunctivitis.
Sweet's syndrome can occur with inflammatory bowel disease, bowel bypass syndrome, and pregnancy. It occasionally occurs as a reaction to certain medications including all-trans retinoic acid and granulocyte colony-stimulating factor.6
Differential diagnosis includes erythema multiforme, deep fungal infection, pyoderma gangrenosum, and cutaneous metastases.
Treatment includes systemic corticosteroids, dapsone, and NSAIDs.
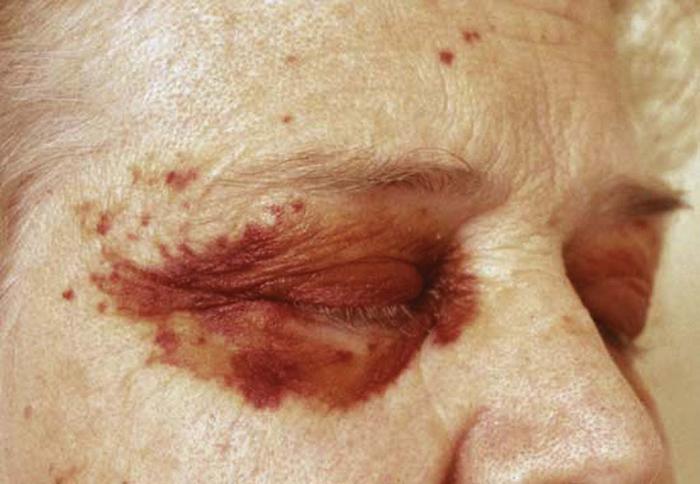
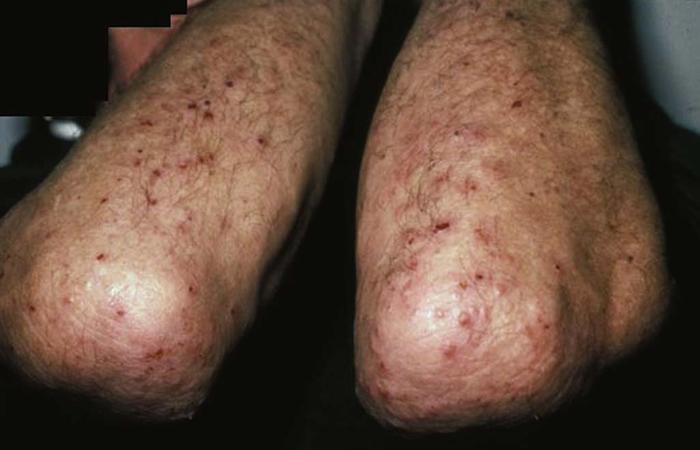
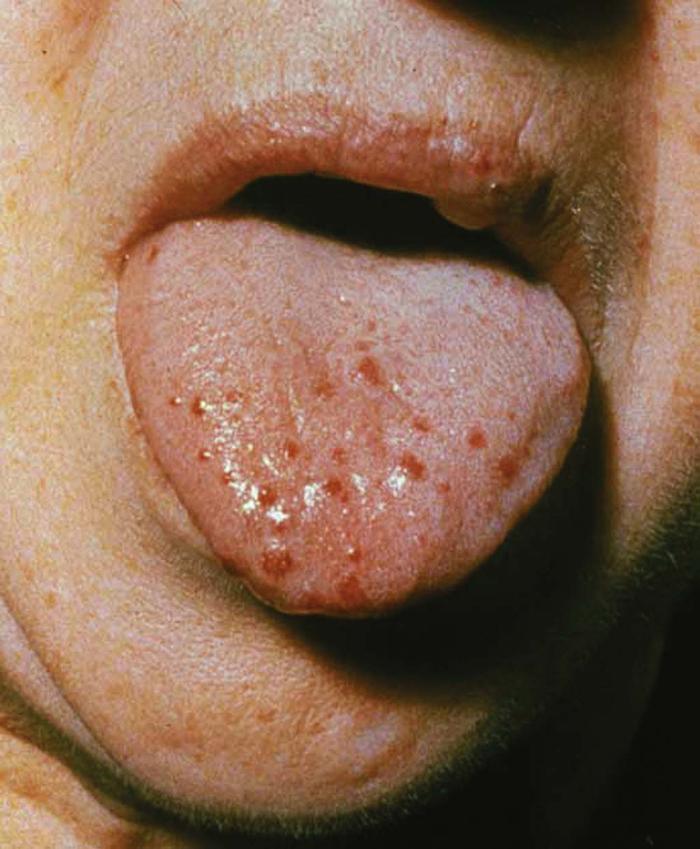
Amyloidosis of the skin may be a sign of multiple myeloma. Affected patients have papules on the eyelids and extremities that become purpuric and ecchymotic due to increased blood vessel fragility secondary to amyloid infiltration of the vessels. The purpura and ecchymosis develop after pressure or rubbing (pinch purpura) (Fig. 14) or may be spontaneous. Many patients have prominent macroglossia. The prognosis for primary systemic amyloidosis is poor.
Differential diagnosis includes nodular amyloidosis.
Treatment includes systemic chemotherapy and stem cell transplantation.
Paraneoplastic pemphigus (Fig. 15), characterized by intractable stomatitis and blisters on the trunk and extremities, has features of pemphigus and erythema multiforme. Direct immunofluorescence reveals deposition of IgG intercellularly and at the dermal-epidermal junction. Paraneoplastic pemphigus has a strong association with non-Hodgkin's lymphoma, chronic lymphocytic leukemia, and Castleman's disease with or without myasthenia gravis. Severe pulmonary disease (bronchiolitis obliterans) with respiratory failure is often the cause of death in affected patients.7
Differential diagnosis includes pemphigus vulgaris, bullous pemphigoid, and erythema multiforme.
Treatment includes treatment of the underlying malignancy, systemic corticosteroids, steroid-sparing immunosuppressants, rituximab, and plasmapheresis.
Erythema gyratum repens is a rare but very distinctive skin disease characterized by reddened concentric bands in a whorled or woodgrain pattern. Affected patients have severe pruritus and peripheral eosinophilia. Erythema gyratum repens has a strong association with lung cancer; the association with breast, cervical, and gastrointestinal cancers is less strong.
Treatment of the underlying malignancy treats the skin disease.
Multiple lentigines occur with LEOPARD syndrome (Fig. 16), an acronym for lentigines, electrocardiographic changes, ocular telorism, pulmonary stenosis, abnormal genitalia, retarded growth, and deafness. Inheritance is autosomal dominant, and many cases occur sporadically.
Carney complex encompasses LAMB syndrome (lentigines, atrial myxoma, mucocutaneous myxomas, and blue nevi) and NAME syndrome (nevi, atrial myxoma, myxoid neurofibromas, and ephelides), entities known to pediatricians, cardiologists, and dermatologists. Recognition of these syndromes is critical because identification and removal of the associated atrial myxomas may be lifesaving. The inheritance pattern for both syndromes is autosomal dominant.
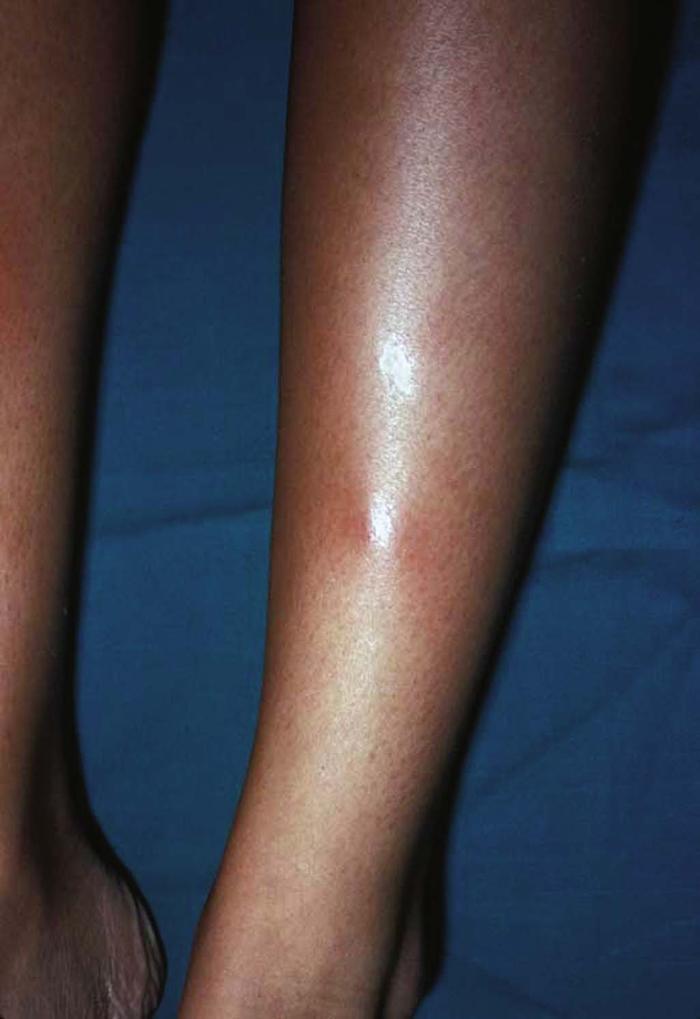
Pseudoxanthoma elasticum (Fig. 17) is characterized by yellow papules over redundant skin folds on the neck, abdomen, and groin, giving the skin the appearance of plucked chicken skin. Pseudoxanthoma elasticum represents a defect in elastic fibers, which become brittle and calcified. Skin biopsy reveals swollen, fragmented elastic fibers, and fundoscopic examination reveals angioid streaks in Bruch's membrane. Associated signs of pseudoxanthoma elasticum include hypertension, peripheral vascular and coronary artery disease, retinal and gastrointestinal hemorrhage, and stroke. Disease is transmitted in a sporadic or inherited (autosomal recessive) fashion. Mutations in the gene ABCC6 on chromosome 16 have been linked to pseudoxanthoma elasticum.
Differential diagnosis includes cutis laxa, Ehlers-Danlos syndrome, and perforating calcific elastosis.
No definitive treatment is available.
Ehlers-Danlos syndrome is a heterogeneous group of connective tissue disorders characterized by joint hyperextensibility, hypermobility, skin and vessel fragility, and fish-mouth scars. Ehlers-Danlos syndrome is characterized by abnormalities in collagen biosynthesis, which can affect many organ systems.
Eleven types of Ehlers-Danlos syndrome have been identified with varying associated features, including mitral valve prolapse, blue sclerae, vascular aneurysm, aortic dissection, hernias, angina, gastrointestinal bleeding (perforation), and peripheral vascular disease. Genetic testing for specific mutations has demonstrated redundancy and has reduced Ehlers-Danlos syndrome from eleven to seven types. Patients with vascular (type IV) Ehlers-Danlos syndrome are prone to arterial rupture and have the highest mortality.
Differential diagnosis includes cutis laxa.
Treatment includes protecting the skin and treating systemic disease.
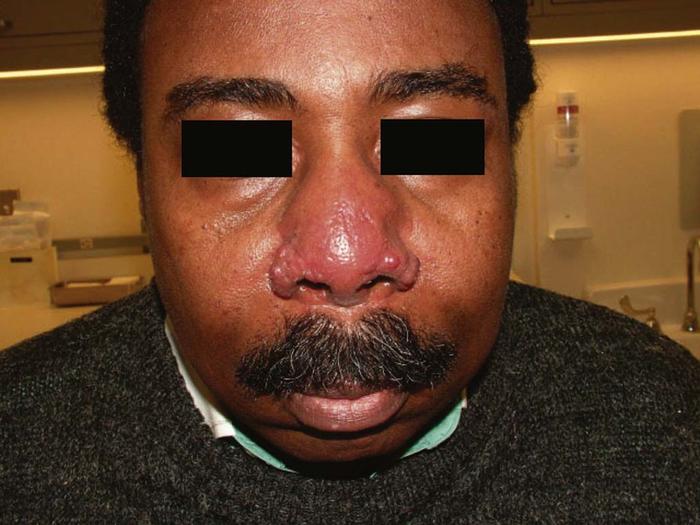
Sarcoidosis is a multisystem, granulomatous disease of the lungs, bones, central nervous system, lymph nodes, eyes, and skin. The disease is more common in women and has a higher prevalence in African Americans. Skin disease, affecting 25% to 35% of patients, includes red to purple indurated plaques of the nose (lupus pernio) (Fig. 18), midfacial papules, annular plaques, and plaques or nodules on the trunk and extremities. Sarcoidal disease also has a predilection for scars. Erythema nodosum, an acute, painful panniculitis that usually affects the shins, is the most common nonspecific cutaneous manifestation of sarcoidosis.
Differential diagnosis includes rosacea, trichoepitheliomas, granulomatous syphilis, and granuloma annulare.
Treatment includes systemic corticosteroids, intralesional corticosteroids for localized disease, methotrexate, thalidomide, antimalarials, and tumor necrosis factor-α (TNF-α) inhibitors.
Psoriatic arthritis (PsA) affects approximately 5% to 10% of patients with psoriasis. Asymmetric fusiform swelling of the distal interphalangeal joints (sausage digits), in association with oligoarthritis and tenosynovitis can be seen in up to 70% of PsA patients. Other presentations include symmetric polyarticular arthritis (15%), distal interphalangeal joint disease with nail damage (16%), arthritis mutilans with erosion of the phalanges (5%), and ankylosing spondylitis (5%). The arthritis can resemble rheumatoid arthritis. Approximately 50% of affected patients have the HLA-B27 genotype.
Differential diagnosis includes osteoarthritis and rheumatoid arthritis.
Treatment includes TNF-α inhibitors, metrotrexate, NSAIDs, and steroid-sparing immunosuppressants.
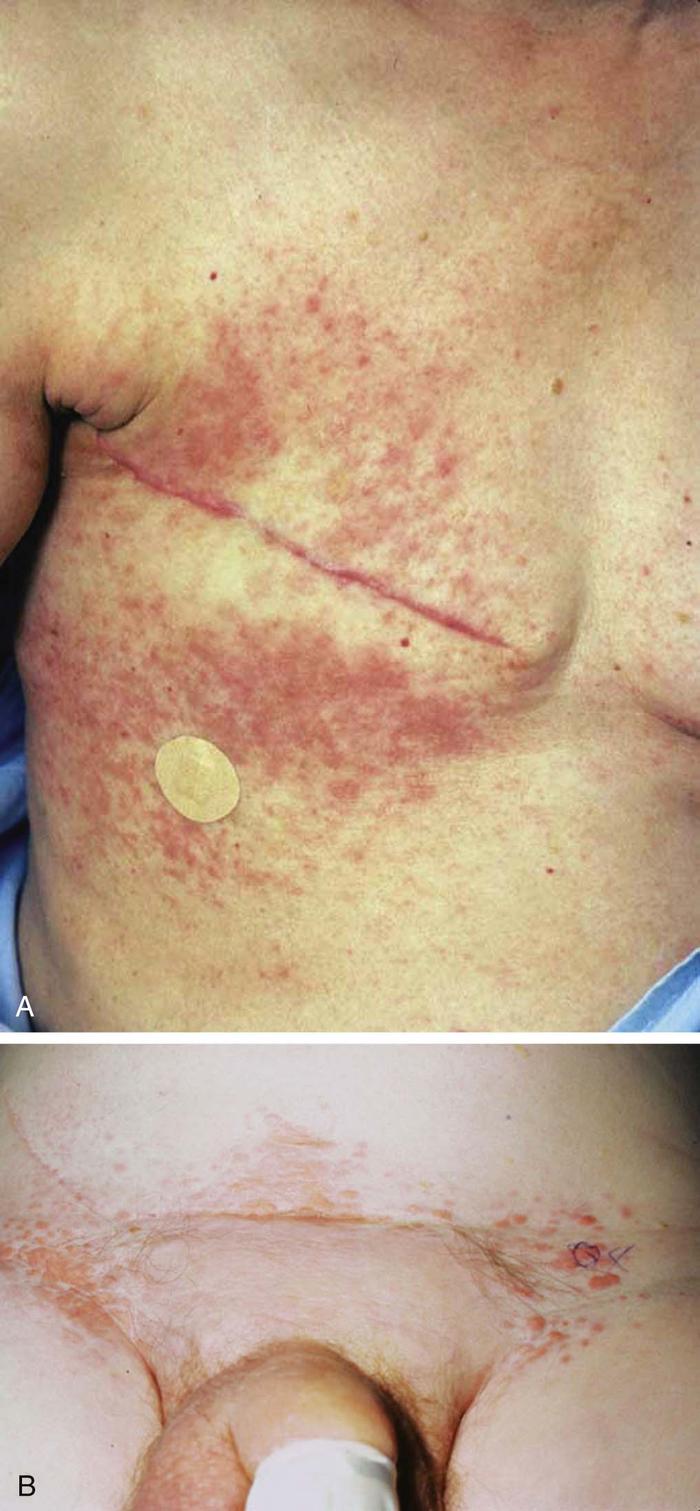
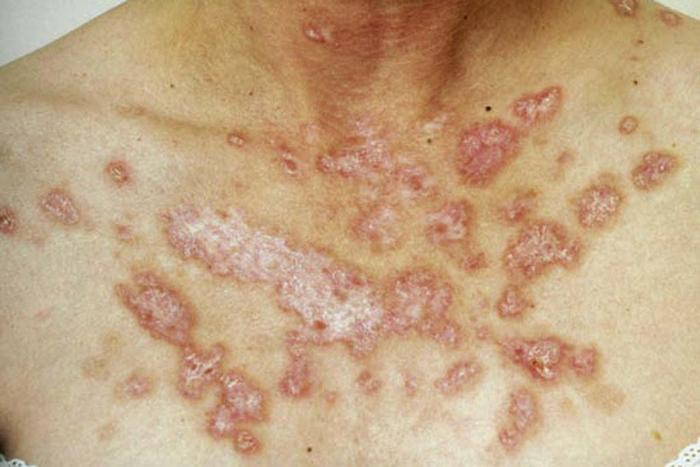
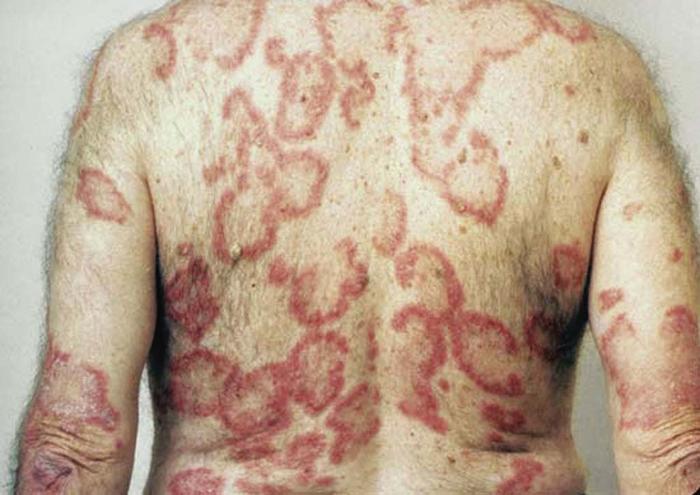
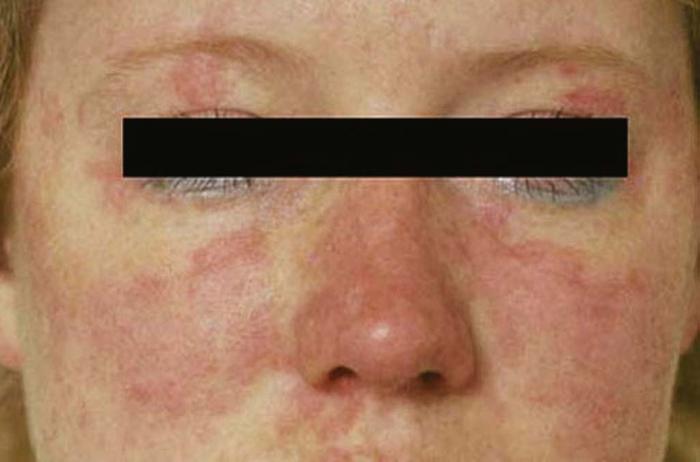
Lupus erythematosus is an autoimmune photosensitive dermatosis that can be localized or systemic, often with significant overlap. Localized cutaneous (discoid) lupus erythematosus (DLE) (Fig. 19), usually localized to the head or neck, is characterized by atrophic, scarring plaques on sun-exposed areas. Five percent of patients develop SLE.
Subacute cutaneous lupus erythematosus (SCLE) (Fig. 20) is characterized by annular pink to red plaques in a sun-exposed, shawl-like distribution on the chest, back, and arms. Unlike DLE, there is no scarring. Serology is often positive in SCLE-antinuclear antibody (80%) and antibodies to Ro/SSA antigen. SCLE patients account for 10% to 15% of the entire lupus erythematosus population.
The cutaneous manifestations of SLE include malar erythema, photosensitivity, oral ulcers, discoid plaques, bullae, purpura, calcinosis cutis, and alopecia. The butterfly rash (malar erythema) is the most common expression of SLE (Fig. 21).
Differential diagnosis includes dermatomyositis, Sjögren's syndrome, photosensitive drug eruption, acute rheumatic fever, and pellagra.
Treatment includes sun protection; intralesional, topical, and systemic corticosteroids; antimalarials; dapsone; and immunosuppressants.
Scleroderma is an autoimmune skin disease that can be localized or generalized. The localized form, known as morphea, begins as erythematous patches that evolve into dusky, hypopigmented, indurated plaques with violaceous borders, usually on the trunk. The systemic or generalized forms are subdivided into CREST syndrome (calcinosis, Raynaud's phenomenon, esophageal dysmotility, sclerodactyly, telangiectasias) and progressive systemic sclerosis. Presence of anticentromere antibodies correlates with CREST syndrome; SCL-70 antibodies correlate with progressive systemic sclerosis. Patients with CREST syndrome have a better prognosis.
Differential diagnosis includes diabetic sclerodema, scleromyxedema, and chronic graft-versus-host disease.
Treatment includes vasodilating drugs, phototherapy (UVA1) for limited disease, methotrexate, and cyclophosphamide.
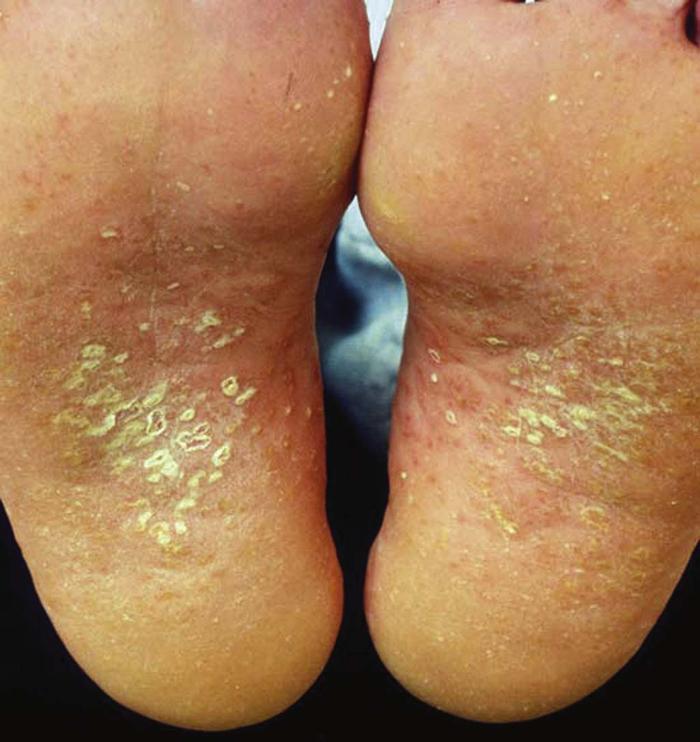
Reactive arthritis (Reiter's syndrome with conjunctivitis, urethritis, and diarrhea) (Fig. 22) usually follows a bout of gastroenteritis or urethritis. Implicated organisms include Campylobacter, Shigella, Salmonella, Ureaplasma, and Yersinia species. Affected patients, usually men, often have vesicles and crusted plaques on the penis (circinate balanitis) and erythematous pustules and papules on the palms and soles (keratoderma blennorrhagicum) that can mimic pustular psoriasis. More than 50% of patients have sacroiliitis, correlating with the presence of HLA-B27 antigen, but few patients have the classic triad of urethritis, conjunctivitis, and arthritis.
Differential diagnosis includes psoriasis, juvenile plantar dermatoses, rheumatoid arthritis, ankylosing spondylitis, and gout.
Treatment includes topical corticosteroids, cyclosporine, or acitretin for refractory disease.
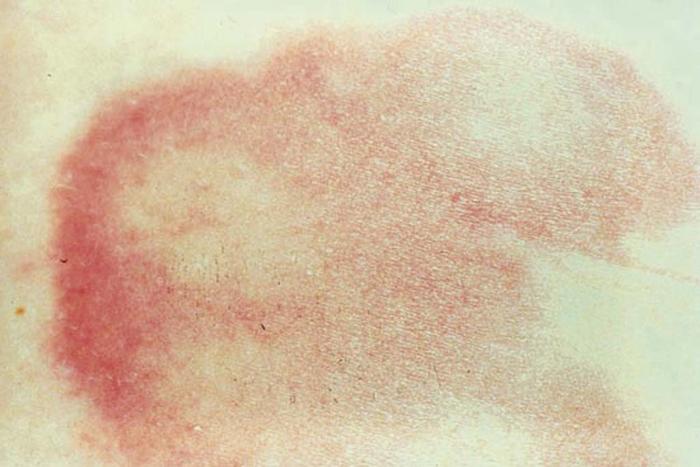
Erythema chronicum migrans, the hallmark of Lyme disease, reflecting early infection with the tick-borne spirochete Borrelia burgdorferi, develops as a red macule or papule at the site of the tick bite and gradually enlarges to an annular, reddened plaque (Fig. 23) that surrounds the bite. Affected patients can have fever, arthralgia, and myalgia, and, occasionally, Bell's palsy. Late sequelae include meningoencephalitis, myocarditis, and peripheral neuropathy. Primary endemic areas in the United States are New England, the upper Midwest, and the Pacific Northwest.
Differential diagnosis includes cellulitis, spider bite, erythema multiforme, and erythema annulare centrifugum.
Treatment is doxycycline.
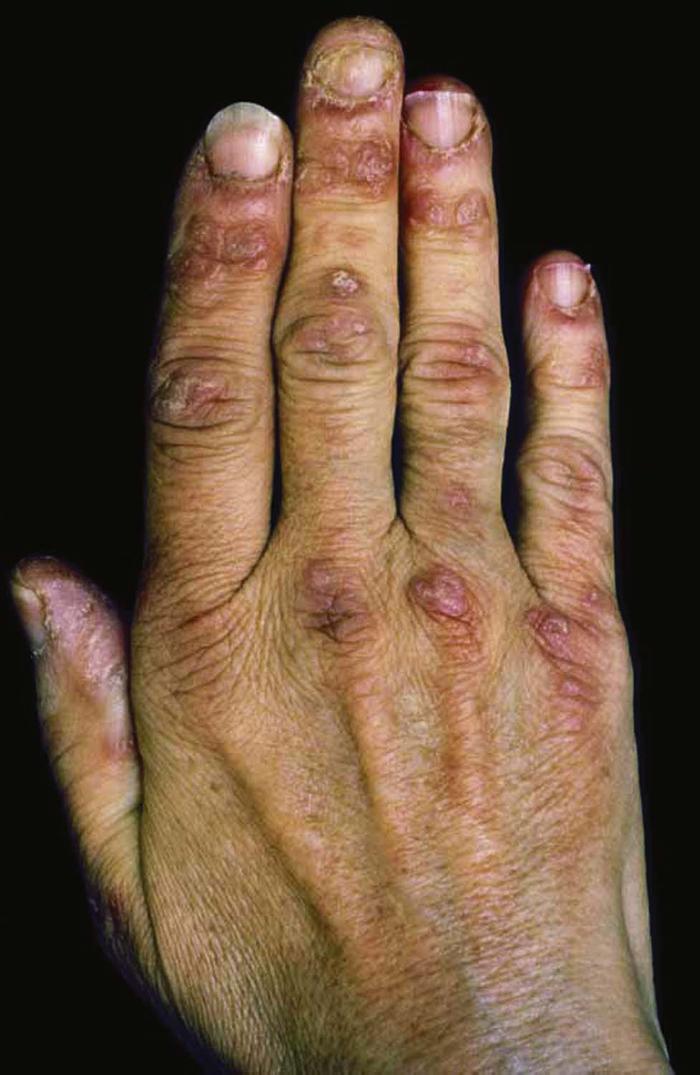
Dermatomyositis, an inflammatory connective tissue disease, is characterized by symmetric proximal muscle weakness (myositis); photosensitivity; papules and plaques on the hands, elbows, and knees (Gottron's papules) (Fig. 24); and periorbital edema with a violaceous hue (heliotrope). Other features include scaly, telangiectatic plaques with atrophy and hypopigmentation (poikiloderma) on the face, neck, trunk, and extremities; malar erythema; and nail abnormalities (periungual telangiectases and cuticular hypertrophy).
Diagnostic criteria include the aforementioned changes plus elevated creatine kinase or aldolase level, positive Jo-1 antibody, and electromyographic changes. In adults, dermatomyositis has a strong association with neoplasm, usually an adenocarcinoma of the breast, gastrointestinal tract, or lung.
Differential diagnosis includes SLE and photosensitive drug eruption.
Treatment includes systemic corticosteroids, methotrexate and other steroid-sparing immunosuppressants, and TNF-α inhibitors.
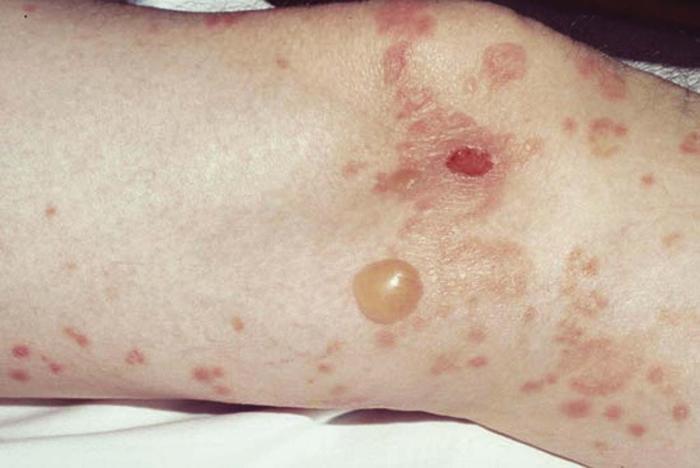
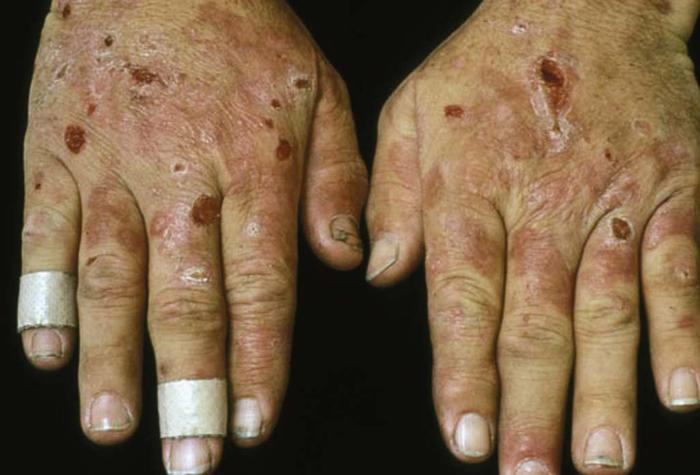
Dermatitis herpetiformis (Fig. 25) is a chronic, intensely pruritic blistering disease characterized by symmetric grouped vesicles, papules, and wheals on the elbows, knees, scalp, and buttocks. Biopsy reveals a characteristic neutrophilic infiltrate, and direct immunofluorescence demonstrates deposition of IgA at the dermal-epidermal junction. Most patients have an asymptomatic gluten-sensitive enteropathy or, less commonly, thyroid disease. Approximately 70% of patients have circulating IgA antibodies against the smooth muscle cell endomysium (antiendomysial antibodies), which are somewhat peculiar to dermatitis herpetiformis.
Differential diagnosis includes linear IgA dermatosis, bullous pemphigoid, scabies, contact dermatitis, and bullous lupus erythematosus.
Treatment includes dapsone, sulfapyridine, and a gluten-free diet.
Acrodermatitis enteropathica is an inherited or acquired condition characterized by pustules, bullae, scaling in an acral and periorificial distribution, and concomitant zinc deficiency. When inherited, acrodermatitis enteropathica results from a mutation in SLC39A, which encodes an intestinal zinc transporter.8 In infants, deficiency can follow breast-feeding, when maternal breast milk contains low levels of zinc. In adults, disease can occur after total parenteral nutrition without adequate zinc supplementation; with alcoholism, other malabsorption states, or inflammatory bowel disease; or as a consequence of bowel surgery. Most patients have diarrhea.
Differential diagnosis includes other nutritional deficiencies, such as niacin or biotin deficiency, and necrolytic migratory erythema.
Treatment is zinc supplementation.
Necrolytic migratory erythema (glucagonoma syndrome) is a rare disease characterized by erythematous, scaly plaques on acral, intertriginous, and periorificial areas, in association with an islet cell tumor of the pancreas. Associated signs include hyperglycemia, diarrhea, weight loss, and atrophic glossitis.
Treatment is rmoval of the tumor.
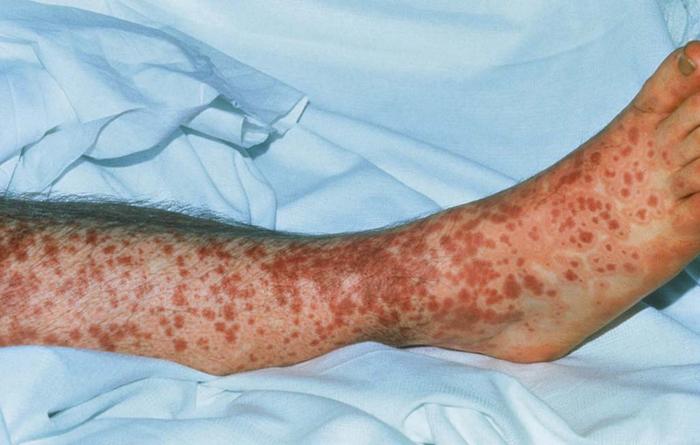
Leukocytoclastic vasculitis (cutaneous small vessel vasculitis) (Fig. 26) associated with circulating type II cryoglobulins, usually yields palpable purpura on the lower extremities. Treatment of the hepatitis C infection often leads to resolution of the vasculitis.
Lichen planus (Fig. 27) is characterized by violaceous, flat, polygonal papules, often on the flexor aspects of the wrists, trunk, medial thighs, genitalia, and oral mucosa. Lichen planus also occurs with primary biliary cirrhosis and hepatitis B virus immunization. Oral erosive lichen planus is the most common expression of lichen planus in hepatitis C patients. Treatment includes topical and intralesional corticosteroids, topical immunomodulators, and phototherapy.
Necrolytic acral erythema, characterized by pruritic keratotic plaques on the upper and lower extremities, is a distinctive finding in hepatitis C infection and can resemble a deficiency dermatosis.
Porphyria cutanea tarda is discussed later.
Gardner's syndrome is an autosomal dominant cancer syndrome characterized by colonic polyposis, osteomas (maxilla, mandible, skull), scoliosis, epidermoid cysts, and soft-tissue tumors (fibromas, desmoids, lipomas). A mutation in the APC gene is responsible for Gardener's syndrome. Adenocarcinoma of the colon develops in 60% of patients by the age of 40 years.
Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome) is an autosomal dominant disorder characterized by numerous telangiectases on the skin and oral mucosa (Fig. 28). Recurrent epistaxis is the most common presenting manifestation of the syndrome, affecting approximately 85% to 90% of patients. Telangiectases can involve the lungs, liver, brain, eyes, and gastrointestinal tract; hemorrhage can occur at any site. Pulmonary arteriovenous fistulae and central nervous system angiomas can also occur.
Differential diagnosis includes generalized essential telangiectasia.
Treatment includes estrogen therapy or oral contraceptives in postpubertal women, laser cauterization, selective embolization, and supportive care.
Muir-Torre syndrome is a disorder characterized by one or more sebaceous tumors (adenoma, epithelioma, carcinoma) and one or more internal neoplasms, usually colorectal or genitourinary, rarely lymphoma. This syndrome results from an inactivating germline mutation of the DNA mismatch repair genes, most often MSH-2. Treatment is isotretinoin and regular GI and genitourinary evaluation.
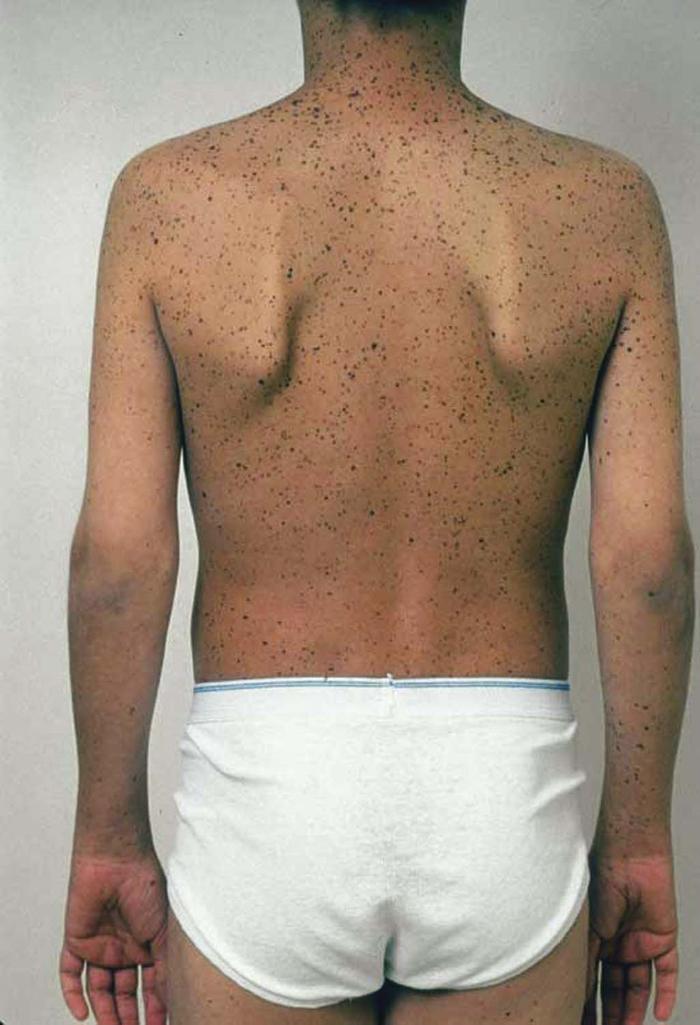
Peutz-Jeghers syndrome is an autosomal dominant disease characterized by lentigines on the skin (periorbital region, dorsal surfaces of the fingers and toes) and mucosa (lips, buccal mucosa) and hamartomas of the stomach, small intestine, and colon. The polyps are usually benign with low malignant potential, but patients have a 10 to 18 times greater lifetime risk of cancer, especially GI malignancies.
Differential diagnosis includes LEOPARD syndrome, Carney complex, and Cronkhite-Canada syndrome.
Treatment includes regular and routine endoscopy and symptomatic treatment for hypogeusia and diarrhea.
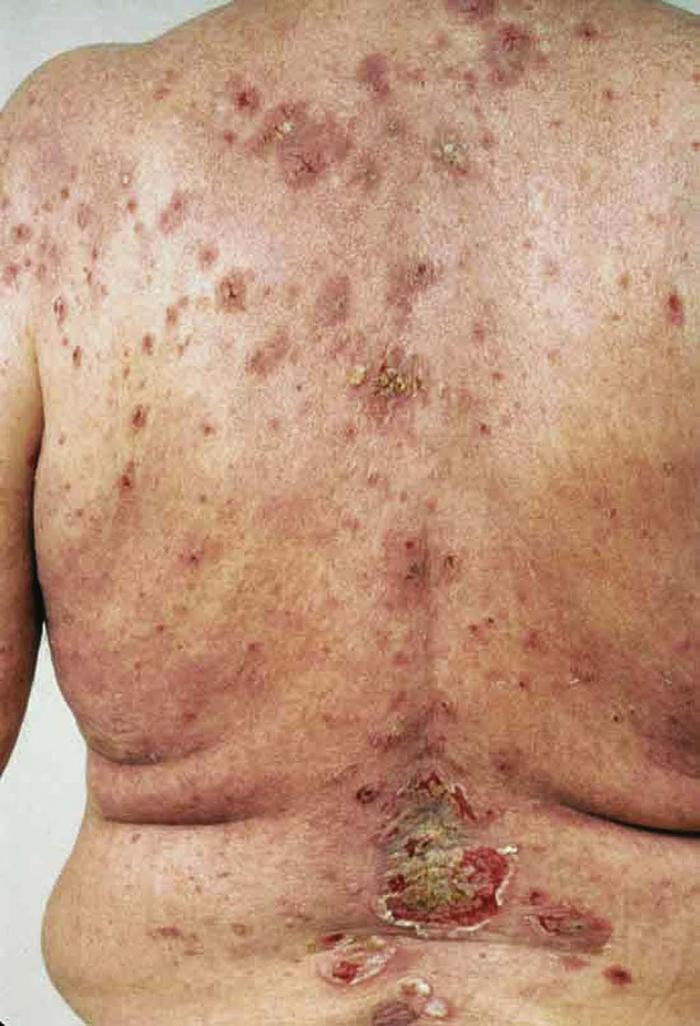
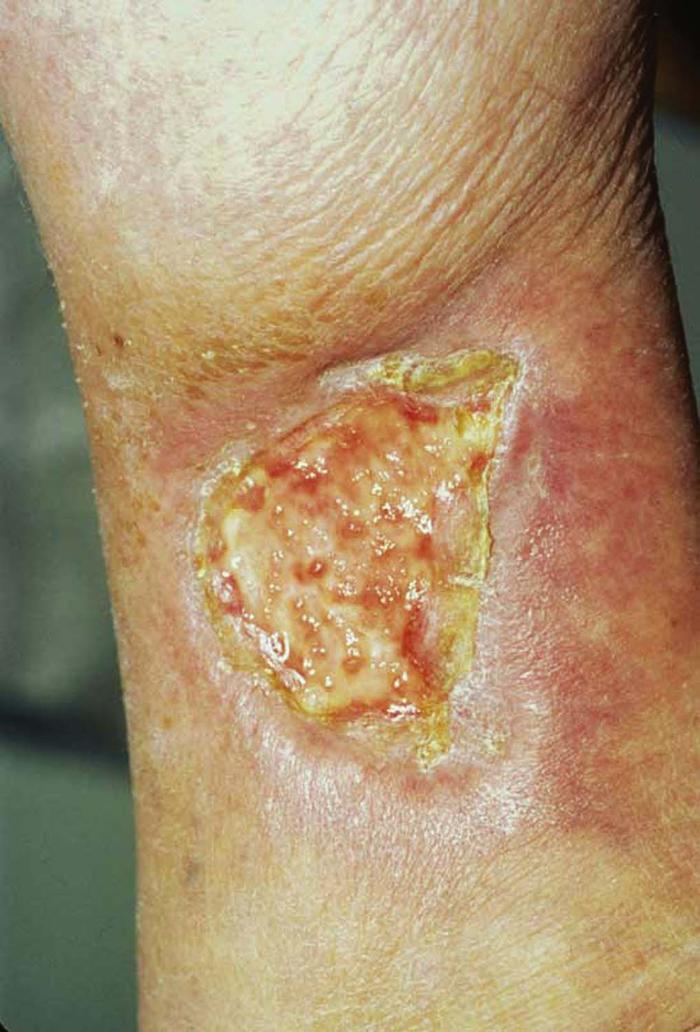
Pyoderma gangrenosum is a neutrophilic dermatosis characterized by painful ulcers with boggy, undermined edges and a border of gray or purple pigmentation (Fig. 29). The ulcers often follow trauma (pathergy) and begin as pustules or nodules that ulcerate and extend centrifugally.9 All body areas may be involved, but the legs are the most common site. Fifty percent of patients have underlying rheumatoid arthritis or inflammatory bowel disease or, less often, a paraproteinemia, usually an IgA gammopathy.
Differential diagnosis includes infection, vasculitis, spider bite, and factitious disorder.
Treatment includes treatment of underlying disease if applicable, local wound care, systemic and intralesional corticosteroids, cyclosporine, and infliximab.
Nephrogenic systemic fibrosis, also known nephrogenic fibrosing dermopathy, is a recently described disorder that resembles scleroderma. Nephrogenic systemic fibrosis occurs in patients who have end-stage renal disease and are on dialysis and occasionally in patients with acute renal failure or after kidney transplantation. Nephrogenic systemic fibrosis is characterized by thick, indurated plaques on the extremities and the trunk. Disease can be progressive, leading to joint contractures. Autopsies have demonstrated that disease is not limited to the skin; visceral organ and muscle fibrosis has been noted. The cause remains unclear, but the MRI contrast agent gadolinium might have a role in the pathogenesis of this condition.10
Differential diagnosis includes scleroderma and scleromyxedema.
Treatment includes immunosuppressive agents, phototherapy, topical steroids, retinoids, and photopheresis, all with little benefit.
Birt-Hogg-Dubé syndrome is a disorder characterized by multiple fibrofolliculomas and trichodiscomas (skin-colored dermal papules on the face and trunk). Patients have a significantly increased risk of renal oncocytoma and chromophobe renal carcinoma. Spontaneous pneumothorax can occur secondary to rupture of pulmonary cysts. Mutations in the folliculin gene on chromosome 17 are responsible for this syndrome.
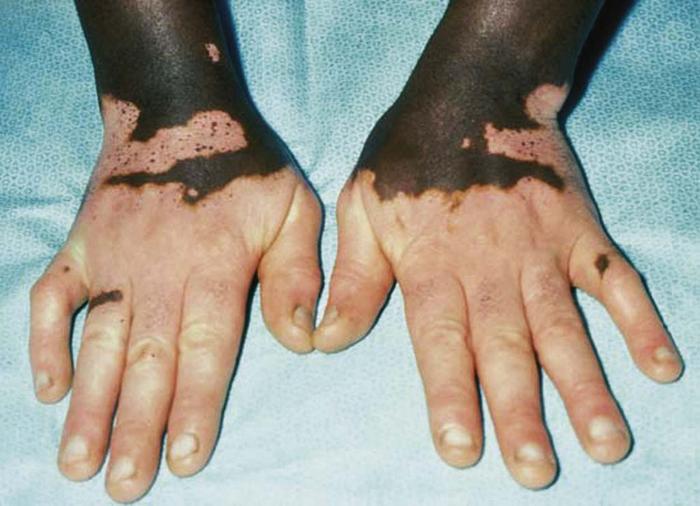
Porphyrias are inherited or acquired disorders of heme biosynthesis and can be erythropoietic, hepatic, or mixed in nature, each associated with a specific enzyme defect in the heme pathway. Porphyria cutanea tarda, the most common porphyria, is a hepatic porphyria with acquired and sporadic forms (Fig. 30). It is caused by a deficiency in uroporphyrinogen decarboxylase, leading to the accumulation of uroporphyrin in the urine and serum.
Precipitating factors include alcohol ingestion, estrogen administration, certain hepatotoxins (dinitrochlorobenzene, carbon tetrachloride), HIV infection, hemochromatosis, and hepatitis C infection. Manifestations of porphyria cutanea tarda include photosensitivity, skin fragility, bullae and erosions on sun-exposed skin (especially dorsal hands), and hypertrichosis. Biopsy reveals a subepidermal bulla with festooning of the dermal papilla. Direct immunofluorescence reveals IgG and C3 at the dermal-epidermal junction and in vessel walls.
Differential diagnosis includes bullous SLE, epidermolysis bullosa acquisita, pseudoporphyria, and variegate porphyria.
Treatment includes phlebotomy and antimalarial drugs.
Pseudoporphyria mimics porphyria cutanea tarda without an enzyme defect; plasma and urinary porphyrins are normal. Medications (NSAIDs [especially naproxen], furosemide, and tetracycline) are the most common cause of pseudoporphyria. Less common causes are tanning bed use and hemodialysis.
Differential diagnosis is the same as for porphyria cutanea tarda.
Treatment includes removal of the cause.
Approximately 30% to 50% of diabetic patients develop skin disease. Box 1 outlines the most common cutaneous manifestations of diabetes, arranged by frequency of occurrence (most to least frequent).
| Box 1: Cutaneous Manifestations of Diabetes Mellitus |
|---|
Diabetic dermopathy (shin spots)
|
Diabetic thick skin
|
Acanthosis nigricans
|
Yellow nails and skin (palms and soles)
|
Acquired perforating disorders
|
Calciphylaxis
|
Necrobiosis lipoidica diabeticorum
|
Diabetic bullae
|
© 2007 Cleveland Clinic Foundation.