Published: August 2010
The term cholestasis originally derives from the Greek and literally means “a standing still of bile.” This disruption of bile flow can occur on a cellular level in the hepatocyte, at the level of the intrahepatic biliary ductules, or from an extrahepatic mechanical obstruction of the bile ducts. Commonly, bile flow is only partially disrupted, giving rise to anicteric cholestasis, or cholestasis without jaundice. Cholestasis is defined, therefore, both clinically and biochemically, with varying degrees of jaundice, pruritus, and elevated levels of conjugated bilirubin, alkaline phosphatase, γ-glutamyl transpeptidase, 5'-nucleotidase, bile acids, and cholesterol. A conventional categorization of cholestatic liver diseases has divided these factors into intrahepatic and extrahepatic causes (Box 1). This chapter discusses the different types of intrahepatic cholestatic liver disease.
| Box 1 Causes of Cholestasis |
|---|
| Intrahepatic Cholestasis |
| Primary biliary cirrhosis |
| Primary sclerosing cholangitis |
| Drugs and toxins |
| Sepsis |
| Malignancy |
| Granulomatous liver disease |
| Intrahepatic cholestasis of pregnancy |
| Hepatitis (viral and alcoholic) |
| Genetic disorders |
| Graft-versus-host disease |
| Post-liver transplantation |
| Extrahepatic Biliary Tract Diseases |
| Choledocholithiasis |
| Bile duct tumors, benign and malignant |
| Ampullary tumors, benign and malignant |
| Pancreatic carcinoma |
| Mirizzi's syndrome |
| AIDS cholangiopathy |
| Parasites |
| Primary sclerosing cholangitis |
Primary biliary cirrhosis (PBC) is a chronic cholestatic liver disease predominantly affecting middle-aged women. It is hypothesized that PBC begins with loss of immune self tolerance, leading to damage of the biliary epithelial cells of small bile ducts. Ongoing immunologic events perpetuate the biliary epithelial cell destruction via direct cytotoxicity or lymphokine-mediated cell damage, leading to disease progression.
PBC is most commonly diagnosed after the age of 40 years. Of patients with PBC, 90% are women. The prevalence is higher in northern European population groups and lower in Japan. Disease prevalence estimates have ranged from 40 to 400 cases per 1,000,000 population, with an incidence between 4 and 30 cases per 1,000,000 per year.1 Recent evidence has suggested that environmental factors, including infectious agents and chemicals, might play a role in inducing PBC in genetically predisposed patients.2
PBC is considered an autoimmune disease, with immune destruction of the interlobular bile ducts resulting in a gradually progressive ductopenia. PBC is generally a progressive disease leading to cirrhosis and death, although there have been reports of prolonged survival, with minimal progression of disease. Patients who are asymptomatic at presentation have a longer survival than those who are symptomatic; however, their survival appears to be shorter than that of an age-matched controlled population. About one third of patients who are asymptomatic at presentation become symptomatic within 5 years. Symptomatic patients have an 8-year survival rate of approximately 50%. Survival models have been developed to predict outcome more precisely and are useful in determining the timing for liver transplantation. If PBC is diagnosed at an early histologic stage and treatment with ursodiol is begun (see later), recent studies have suggested that the long-term survival approaches that of a healthy control population. Patients with more advanced histologic disease at diagnosis, however, have 30% and 50% rates of requiring liver transplantation or death over 10 and 20 years, respectively, despite treatment.3
With the ready availability of automated blood chemistry testing, many cases are diagnosed in an asymptomatic phase. The most common initial symptom is fatigue, which occurs in approximately 70% of patients. Fatigue does not necessarily correlate with the severity of disease, and there is evidence suggesting central nervous system mediation of this symptom. Fatigue severity in PBC has been associated with excessive daytime somnolence, autonomic dysfunction, cognitive impairment, and depression.4-6 Pruritus is also a common symptom, occurring in 50% to 60% of patients. As the disease progresses, patients can develop symptoms of portal hypertension, such as variceal hemorrhage and ascites. Xanthomata, particularly around the eyes (xanthelasma), are commonly found in patients with PBC. PBC is also associated with metabolic bone disease, resulting in premature osteoporosis.
As the disease progresses, there can be fat-soluble vitamin malabsorption caused by a decrease in the biliary secretion of bile acids. There is an increased frequency of other autoimmune disorders in patients with PBC, including autoimmune thyroid disease, sicca syndrome, CREST syndrome (calcinosis, Raynaud’s phenomenon, esophageal dysmotility, and telangiectasia), celiac disease, and inflammatory bowel disease.
The diagnosis of PBC is based on a combination of findings, including cholestatic liver enzyme levels, positive antimitochondrial antibody (AMA), and characteristic liver biopsy findings. An elevated serum alkaline phosphatase level of liver origin is the most common laboratory finding.
The most characteristic laboratory finding in PBC is the presence of the AMA, generally in a titer of 1 : 40 or higher. More than 95% of patients with PBC have a positive AMA. A confident diagnosis of PBC may be made in cases with typical clinical presentation of PBC in the setting of a positive AMA (≥1 : 40), and a cholestatic pattern of liver enzymes with alkaline phosphatase at least 1.5 times the upper limit of normal and AST less than five times the upper limit of normal without the obligation to perform a liver biopsy.7, 8 A liver biopsy should be performed in atypical cases, in cases where an alternative diagnosis is suspected, and to obtain staging information. The liver biopsy findings include portal hepatitis, with granulomatous destruction of bile ducts. The histologic changes are divided into four stages, ranging from stage 1, characterized by portal inflammation and bile duct destruction, through stage 4, characterized by histologic cirrhosis. Overlapping stages can be found in individual patients.
A subgroup of patients have a positive AMA with normal liver enzyme levels. Most of these patients ultimately develop biochemical evidence of cholestasis and symptomatic disease. Another subgroup, with cholestasis and histology suggesting PBC, are AMA negative (AMA-negative PBC).9 The natural history of AMA-positive and AMA-negative PBC appears to be similar. A positive AMA, usually in low titer, can be seen in patients with other autoimmune disorders.10
Treatment of PBC is directed at both the underlying disease and its complications. Ursodeoxycholic acid (UDCA) is a dihydroxy bile acid that is hydrophilic and nonhepatotoxic. Several large randomized trials using UDCA have shown biochemical improvement.11-15 Although controversy exists regarding the effect of UDCA in survival, a survival benefit is demonstrated when only trials with adequate UDCA dosing (13-15 mg/kg/day) and sufficient duration of follow-up are considered. Patients without established cirrhosis at the time of starting UDCA therapy, and those with significant improvement of their alkaline phosphatase with therapy, seem to show the greatest survival benefit. In this regard, the survival of noncirrhotic patients with PBC on UDCA therapy appears similar to that of the general population.16 Similarly, patients with an adequate biochemical response with UDCA therapy also have survival similar to that of the general population.17 UDCA is currently the recommended treatment at a dosage of 13 to 15 mg/kg daily, either in divided doses or as a single daily dose.8
Other drugs have recently been considered for treatment. Methotrexate in combination with UDCA has been shown not to improve the course of PBC compared with UDCA alone in a large randomized trial.18 Oral budesonide seems to improve hepatic histology in PBC, but its role in treatment remains undetermined. Liver transplantation is recommended for patients with decompensated liver disease.
The most common symptom of PBC requiring treatment is pruritus. First-line treatment consists of cholestyramine at a dosage of 4 g/day, up to 16 g daily. At least 4 hours should elapse between taking cholestyramine and any other medication. Rifampicin 300 to 600 mg/day is second-line treatment for pruritus in patients who do not respond to cholestyramine. Opioid antagonists such as naltrexone have also been used in treatment-resistant cases, as has plasmapheresis. Unfortunately, there is no therapy proved to be of benefit for fatigue in PBC.
Patients with stage 4 PBC can develop portal hypertension and should be screened for the presence of esophageal varices when PBC is first diagnosed and every 3 years thereafter. If prominent varices are found, consider primary prophylaxis (pharmacologic or endoscopic). Bone mineral density should be assessed at the time of diagnosis and periodically thereafter. If osteoporosis is present, consider treatment with a bisphosphonate.19 Fat-soluble vitamin deficiency should be considered and screened for in patients with hyperbilirubinemia, and oral replacement may be necessary. The association of thyroid disease with PBC has led to the recommendation of checking the serum thyroid-stimulating hormone level at the time of diagnosis and periodically thereafter. Hypercholesterolemia is commonly seen in PBC, but it has not been demonstrated that this is associated with increased cardiovascular risk. Hypercholesterolemia in these patients should be managed based on each patient’s cardiovascular risk profile.
Guidelines on the management of primary biliary cirrhosis have been published and are available online .8, 20 This is a comprehensive review of PBC with discussions of diagnosis, clinical manifestations, associated conditions, and therapy. Recommendations made in the review are judged on the quality of evidence in the medical literature used to formulate each guideline.
Primary sclerosing cholangitis (PSC) is a chronic, progressive, cholestatic liver disease resulting from inflammation, fibrosis, and destruction of the intrahepatic and extrahepatic bile ducts. This leads to multiple areas of stricturing in the biliary tree and eventually to cirrhosis.
The estimated prevalence of PSC is 60 to 80 cases per 1 million population. There is a 2 : 1 male predominance. Approximately 80% of patients with PSC have inflammatory bowel disease, more commonly ulcerative colitis than Crohn’s disease.21
The pathophysiology of PSC is unclear, but there is evidence suggesting an autoimmune component to the disease. There is also a genetic predisposition, with an increased prevalence of HLA-B2 and DR3 in patients with PSC. Other proposed causes include chronic portal bacteremia, cytotoxic bile acids, and viral infections. The periductal inflammation leads to progressive multifocal stricturing of the intrahepatic and extrahepatic biliary tree.21
PSC is a progressive disease, often leading to biliary cirrhosis within 10 to 15 years. Patients who are asymptomatic at the time of diagnosis fare better than those who are symptomatic, but the disease tends to progress in either case. The average overall survival time is approximately 10 years from the date of diagnosis.
Cholangiocarcinoma is a dreaded complication of PSC, occurring in 4% to 20% of patients; the incidence is even higher in autopsy studies. The development of cholangiocarcinoma is often accompanied by clinical decline but can be difficult to diagnose, even when it is suspected, because of the low sensitivity of biliary brush cytology in this setting. Survival after the diagnosis of cholangiocarcinoma is poor, and cholangiocarcinoma is often considered a contraindication to liver transplantation. Some centers have had favorable outcomes with liver transplantation preceded by radiation and chemotherapy.22
It is common for patients with PSC to be asymptomatic. In one large study, only 56% of patients had one or more symptoms at the time of initial diagnosis.23 The most common symptom is fatigue, which is nonspecific. Other, less-common symptoms include pruritus, weight loss, and fever. Occasionally, patients present with symptoms of portal hypertension, including the onset of ascites or variceal bleeding, or symptoms of bacterial cholangitis. Physical examination at initial presentation may be normal, although jaundice and hepatosplenomegaly are present in up to 50% of patients.
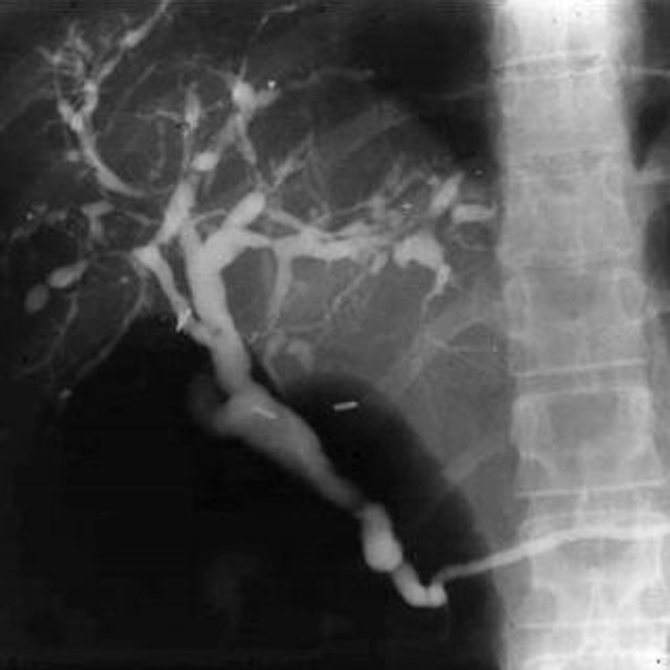
The diagnostic test of choice for PSC is cholangiography, typically endoscopic retrograde cholangiography (ERC). Occasionally, percutaneous transhepatic cholangiography is necessary to establish the diagnosis when ERC is unsuccessful. The cholangiogram typically shows multiple strictures of the intrahepatic and extrahepatic biliary tree (Fig. 1). In one large study, 27% of patients had intrahepatic ductal involvement only and 6% had only extrahepatic ductal changes.23 Magnetic resonance cholangiography (MRC) has also been used in the diagnosis of PSC. When compared with ERC in one study, MRC had a sensitivity of 85% to 88% and a specific of 92% to 97%, with good interobserver agreement.24 It remains to be seen whether MRC will surpass ERC as the first-line test for the diagnosis of PSC.
Liver biopsy is not diagnostic for PSC, but findings often include commonly the absence of intralobular bile ducts (ductopenia), bile duct proliferation, and periductal fibrosis, with an onion-skin fibrosis and nodular fibrous scars. A liver biopsy is often not necessary for the routine diagnosis of PSC.25 Liver enzyme studies typically show an elevated alkaline phosphatase level of biliary origin, although there is a subgroup of patients with early PSC who present with a normal alkaline phosphatase level.
“Currently, no medical therapy has been shown to be beneficial in PSC. A 2-year randomized, controlled trial using UDCA at a dose of 12 to 15 mg/kg/d in patients with PSC was associated with improved liver tests, however there was no beneficial effect on survival, liver histology, cholangiographic appearance or symptoms.26 Subsequently, studies testing higher doses of UDCA (between 17 and 23 mg/kg/d) showed trends towards improved survival but did not reach statistical significance. Recently, a 5-year randomized controlled trial of UCDA at 28 to 30 mg/kg/d demonstrated that high dose UDCA was associated with improved liver tests but did not improve survival and was associated with higher rates of serious adverse events. Based on these results, high dose UDCA (25 to 30 mg/kg/d) cannot be recommended in patients with PSC. Consideration of medical therapy for patients with PSC in the setting of prospective studies is reasonable, but there is not treatment that can be recommended at this time”. Medical management of PSC is therefore limited to complications that arise during the course of the disease. Most of these complications and treatments are similar to those listed earlier for the management of PBC.
Up to 20% of patients with PSC develop jaundice, cholangitis, or both, caused by a dominant stricture of the biliary tree, which can be treated with balloon dilation with or without the placement of a biliary stent. This is usually done endoscopically, but it can be done percutaneously. Although there are no established guidelines for surveillance for cholangiocarcinoma in patients with PSC, a high index of suspicion should be maintained.
Liver transplantation is effective for patients who have evidence of end-stage liver disease or who have recurrent bouts of cholangitis that cannot be controlled with dilation of a dominant stricture. Unfortunately, PSC recurs in 15% to 20% of cases, and recurrence is often associated with loss of the graft.
Drugs are a common cause of cholestasis. The spectrum of drug-induced liver injury can range from acute reversible cholestasis to chronic cholestasis with loss of bile ducts. In a large study of 1100 cases, acute cholestasis accounted for about 17% of liver-related adverse drug reactions.30 Drugs can interfere with various stages of bile acid metabolism, including uptake, transport, and secretion at the hepatocyte level.31
Drug-induced cholestasis can be categorized into acute and chronic forms (Box 2).32 The acute forms are subdivided into cholestasis without inflammation (bland cholestasis), cholestasis with inflammation, and cholestasis with bile duct injury. Chronic forms include a vanishing bile duct syndrome and a sclerosing cholangitis-like syndrome.
| Box 2 Drug-Induced Cholestasis |
|---|
| Cholestasis without Hepatitis |
| Estrogens |
| Anabolic steroids |
| Cyclosporine |
| Tamoxifen |
| Azathioprine |
| Cholestasis with Hepatitis |
| Chlorpromazine |
| Macrolide antibiotics |
| Tricyclic antidepressants |
| Carbamazepine |
| Amoxicillin-clavulanate |
| Oxypenicillins |
| Nonsteroidal anti-inflammatory drugs |
| Azathioprine |
| Cholestasis with Bile Duct Injury |
| Dextropropoxyphene |
| Flucoxacillin (floxacillin) |
| Carmustine |
| Toxins: paraquat, methylenedianiline |
| Vanishing Bile Duct Syndrome |
| Chlorpromazine |
| Flucloxacillin (floxacillin) and other oxypenicillins |
| Amoxicillin-clavulanic acid |
| Ampicillin |
| Amitriptyline |
| Azathioprine |
| Barbiturates |
| Carbamazepine |
| Chlorothiazide |
| Cotrimoxazole |
| Clindamycin |
| Chlorpromazine |
| Cimetidine |
| Cyproheptadine |
| Dicloxacillin |
| Erythromycin esters |
| Estradiol |
| Flucloxacillin |
| Glycyrrhiza |
| Haloperidol |
| Ibuprofen |
| Imipramine |
| Methyltestosterone |
| Norandrostenolone |
| d-Penicillamine |
| Phenytoin |
| Prochlorperazine |
| Tetracycline |
| Terbinafine |
| Thiabendazole |
| Tiopronin |
| Tolbutamide |
| Sclerosing Cholangitis-like Syndrome |
| Floxuridine |
Intralesional and scolicidal agents
|
Data from Chitturi S, Farrell GC: Drug-induced cholestasis. Semin Gastrointest Dis 2001;12:113-124.
Drug-induced cholestasis can be accompanied by nausea, anorexia, malaise, and pruritus.32 Symptoms can occur weeks to months after beginning treatment.
Drugs that cause cholestasis with bile duct injury often are accompanied by additional clinical features, such as fever, rigors, jaundice, and tender hepatomegaly mimicking acute cholangitis. Drugs that result in a vanishing bile duct syndrome can lead to progressive cholestasis, with prolonged jaundice, pruritus, and, occasionally, cirrhosis and liver failure.
The most important tool in the diagnosis of drug-induced cholestasis is a careful medical history, eliciting a history of taking prescribed, over-the-counter, or alternative medications, including herbs. Biliary obstruction should be excluded with an imaging study, ultrasound, or computed tomography (CT) of the biliary tree. The mainstay of treatment is withdrawal of the drug. Management of symptoms associated with cholestasis are similar to those for PBC.
Most cholestatic hepatic injury resolves with withdrawal of the offending medication. A small subgroup of patients develop progressive liver disease, resulting in biliary cirrhosis and liver failure.
Intrahepatic cholestasis is often seen in patients who have sepsis.33 Circulatory endotoxins associated with sepsis induce cytokine production, including tumor necrosis factor α, interleukin-1, and interleukin 6, which results in impaired bile acid transport. The cholestasis of infection is often seen in severely ill hospitalized patients, often in the intensive care unit (ICU). Other factors can contribute to the cholestasis, including medications and total parenteral nutrition. Calculous or acalculous cholecystitis or biliary obstruction is often a concern in this setting. Ultrasound can be a helpful diagnostic tool in this circumstance. Ultrasound is noninvasive and can be done in the ICU. Therapy for sepsis-induced cholestasis consists of treating the underlying infection. Outcomes usually are dictated more by the patient’s underlying disease than by the cholestasis itself.
Primary liver cancer—hepatocellular carcinoma—and metastatic cancer are associated with a liver enzyme pattern suggestive of cholestasis. They are more properly categorized as infiltrative disorders but are discussed here because of their similarity to cholestatic diseases. Hepatocellular carcinoma, once a relatively uncommon tumor, has been increasing in incidence since the 1990s because of its association with hepatitis C–induced cirrhosis. In this setting, the estimated incidence of the development of hepatocellular carcinoma is 1% to 4% per year.34 Cirrhosis from causes other than hepatitis C, particularly hepatitis B and hemochromatosis, is also associated with the development of hepatocellular carcinoma. Hepatocellular carcinoma is often suspected in patients who have previously stable cirrhosis and who have experienced a precipitous clinical decline without other explanation. The diagnosis is made by abdominal imaging techniques, including ultrasound, CT, and magnetic resonance imaging (MRI).
Therapeutic approaches to hepatocellular carcinoma include surgical resection, liver transplantation, and techniques designed to shrink the tumor, such as alcohol injection or radiofrequency ablation.
Metastatic carcinoma can also manifest with cholestasis. The hepatic component is usually found after the diagnosis of carcinoma is made, although it is occasionally the presenting feature. Cholestasis can also occur in patients as a paraneoplastic syndrome in the absence of metastatic disease to the liver. This nonmetastatic cholestasis has been described in non-Hodgkin’s lymphoma, prostate cancer, and renal cell carcinoma.35-38
Granulomatous liver diseases are more accurately classified as infiltrative diseases but are discussed here because the pattern of liver enzyme abnormality resembles that seen with cholestasis. Granuloma formation in the liver occurs in various disorders, including systemic infections from bacteria, viruses, fungi, rickettsia, spirochetes, and parasites; drugs and chemicals; immune-mediated diseases, such as sarcoidosis and primary biliary cirrhosis; and neoplasms, such as Hodgkin’s disease (Box 3).39, 40 The list of commonly used drugs that result in hepatic granulomas is extensive; it includes allopurinol, quinidine, sulfonamides, and sulfonylureas. The finding of granulomas on liver biopsy is often expected, for example, in patients with suspected primary biliary cirrhosis who present with cholestasis and a positive mitochondrial antibody or patients with known sarcoidosis who present with cholestasis. On the other hand, when granulomas are found on liver biopsy unexpectedly or as part of an evaluation for a systemic illness, a thorough investigation should be undertaken to look for the underlying cause.
| Box 3 Causes of Hepatic Granulomatous Liver Diseases |
|---|
| Chemicals |
| Beryllium |
| Drugs |
| Allopurinol |
| Carbamazepine |
| Chlorpropamide |
| Hydralazine |
| Methyldopa |
| Nitrofurantoin |
| Phenytoin |
| Procainamide |
| Quinidine |
| Sulfonamides |
| Sulfonylureas |
| Infection |
| Bacteria |
| Brucellosis |
| Tularemia |
| Yersinia |
| Propionibacterium |
| Pseudomonas pseudomallei |
| Spirochetes: Treponema |
| Rickettsia: Q fever |
| Fungi |
| Histoplasmosis |
| Coccidiomycosis |
| Blastomycosis |
| Aspergillus |
| Actinomycosis |
| Nocardia |
| Cryptococcus |
| Candida |
| Mycobacteria |
| Tuberculosis |
| Atypical mycobacteria |
| Leprosy |
| Parasites |
| Ascaris |
| Toxocara |
| Schistosoma |
| Leishmania |
| Viruses |
| Epstein-Barr Virus |
| Cytomegalovirus |
| HIV |
| Miscellaneous |
| Sarcoidosis |
| Primary biliary cirrhosis |
| Hodgkin's disease |
| Non-Hodgkin's lymphoma |
| Inflammatory bowel disease |
| Systemic lupus erythematosus |
| Whipple's disease |
| Wegener's granulomatosis |
| Talc in drug abusers |
Data from Guckian JC, Perry JE: Granulomatous hepatitis. An analysis of 63 cases and review of the literature. Ann Intern Med 1966;65:1081-1100; Cunningham D, Mills PR, Quigley EM, et al: Hepatic granulomas: Experience over a 10-year period in the West of Scotland. Q J Med 1982;202:162-170.
Evaluation should begin with a careful history including, for example, risk factors for HIV, exposure to tuberculosis, or exposure to farm animals, which presents a risk for brucellosis and Q fever.
Because exposure to drugs is a common cause of granulomatous liver disease, a history of medication use is essential. Further diagnostic testing is often necessary to ascertain the cause; this should include chest x-ray; serologic evaluation for fungi, Brucella, Treponema, HIV, other viruses, and mitochondrial antibody and angiotensin-converting enzyme levels; tuberculin skin testing; and special stains of the liver biopsy for fungus and acid-fast bacilli (AFB). More-extensive evaluation, such as abdominal or chest CT scanning, may be necessary if lymphoma is suspected.
Treatment of granulomatous liver disease is disease specific. It may be as simple as stopping an offending drug. A trial of corticosteroids in patients with idiopathic granulomatous hepatitis who are symptomatic, with fever, myalgias, and arthralgias, may be helpful. Empirical antituberculous therapy should be considered before instituting corticosteroids.
Intrahepatic cholestasis of pregnancy (ICP) can occur in the second or third trimester. There appears to be a genetic component because it has been reported to occur in family members.41 It is likely that hyperestrogenemia associated with a pregnancy plays a role.42 The altered metabolism of progesterone has also been implicated. The hallmark clinical feature of intrahepatic cholestasis of pregnancy is pruritus. Jaundice can occur, and laboratory findings reveal the typical features of cholestasis, including elevated levels of serum bile acids, alkaline phosphatase, and total bilirubin. UDCA has been used in ICP to relieve pruritus, and it appears to be safe for mother and fetus.43 Symptoms resolve within several days of delivery but can recur during subsequent pregnancies.
Occasionally, viral hepatitis manifests with signs and symptoms of cholestasis characterized by jaundice and pruritus. The clinical course can last for several months.44
Alcoholic hepatitis generally manifests with features of cholestasis. It is often accompanied by fever, and the clinical presentation can be confused with that of cholangitis. A careful medical history is essential to confirm a history of ethanol abuse or dependency.
Rare syndromes result from mutations of genes responsible for transporting biliary constituents from the space of Disse across the basal lateral (sinusoidal) membrane and across the canalicular membrane into the bile duct. Transporter gene mutations can result in hereditary cholestasis and include such disorders as Byler’s disease and benign recurrent intrahepatic cholestasis.45 Byler’s disease is characterized by cholestasis occurring early in life that progresses to cirrhosis and death, usually in early childhood. Benign recurrent intrahepatic cholestasis is characterized by episodic jaundice and pruritus lasting for several weeks to months, with long symptom-free intervals. The disease does not progress to cirrhosis. Multiple family members can be affected. Cystic fibrosis can result in cholestasis caused by gene mutations at the level of the bile duct, resulting in inspissated bile.46
Graft-versus-host disease (GVHD) can be seen within the first 100 days after bone marrow transplantation (acute GVHD) or after that time (chronic GVHD). It occurs in up to 50% of patients after bone marrow transplantation and is believed to be caused by T cells of the donor marrow reacting against host antigens, resulting in cytokine damage of the affected organ. GVHD can affect the skin, liver, and gastrointestinal tract.
Although hepatic involvement is usually associated with cholestatic liver enzyme levels, other causes of cholestasis are common in this patient population. GVHD often has to be distinguished from viral infections, drug toxicity, and hepatic veno-occlusive disease. Liver biopsy provides the most definitive way to distinguish the various causes of cholestasis in this patient population. Treatment of GVHD consists of prophylactic measures and treatment of active disease. The most common prophylactic regimen is a combination of methotrexate and cyclosporine. Various treatment regimens of acute of GVHD have been used, including corticosteroids, antithymocyte globulin, tacrolimus, and mycophenolate.47 Chronic GVHD has also been treated with various agents, including prednisone, cyclosporine, thalidomide, psoralen, ultraviolet irradiation, UDCA, tacrolimus, rapamycin, and mycophenolate.48 Less than 50% of patients treated for GVHD sustain a cure. In general, the more severe the skin, liver, or gut involvement, the less favorable the outcome.
Total parenteral nutrition (TPN) is associated with liver dysfunction resulting in steatosis, cholestasis, and cirrhosis. Intrahepatic cholestasis can occur after 2 to 3 weeks of TPN therapy and is associated with elevations in serum bilirubin and alkaline phosphatase levels.
Cholestasis usually reverses after TPN is stopped but is of concern if the patient requires long-term TPN. Progressive liver disease, including cirrhosis, may be associated with long-term TPN.49
Cholestasis is often seen after liver transplantation and is caused by various conditions (Box 4).50 In the first few months after transplantation, cholestasis is often associated with bacterial infections or viral infections, particularly cytomegalovirus. Medications, both antibiotics and immunosuppressive drugs typically used after transplantation, are also associated with cholestasis. Acute cellular rejection is often heralded by the onset of abnormalities in cholestatic liver enzyme levels. Later after transplantation, other causes of cholestasis are more common, including chronic rejection, fibrosing cholestatic hepatitis resulting from recurrent hepatitis B or C, or a recurrence of the original disease, such as PBC or PSC.
| Box 4 Causes of Cholestasis After Liver Transplantation |
|---|
| Infections (bacterial, cytomegalovirus) |
| Medications (immunosuppressive drugs, antibiotics) |
| Viral hepatitis B and C |
| Rejections, acute and chronic |
| Recurrence of disease (primary biliary cirrhosis, primary sclerosing cholangitis) |