Published: April 2013
Last Reviewed: August 2017
Ascites is defined as the accumulation of fluid in the peritoneal cavity. It is a common clinical finding, with various extraperitoneal and peritoneal causes (Box 1), but it most often results from liver cirrhosis. The development of ascites in a cirrhotic patient generally heralds deterioration in clinical status and portends a poor prognosis.
| Extraperitoneal Causes |
|---|
| Budd-Chiari syndrome |
| Chylous ascites |
| Cirrhosis |
| Congestive heart failure |
Hypoalbuminemia
|
| Myxedema |
| Pancreatitis |
| Peritoneal Causes |
| Endometriosis |
Infection
|
Malignancy
|
| Other |
Ascites is the most common major complication of cirrhosis and is an important landmark in the natural history of chronic liver disease. If observed for 10 years, approximately 60% of patients with cirrhosis develop ascites requiring therapy.
Cirrhotic ascites forms as the result of a particular sequence of events. Development of portal hypertension is the first abnormality to occur. As portal hypertension develops, vasodilators are locally released. These vasodilators affect the splanchnic arteries and thereby decrease the effective arterial blood flow and arterial pressures. The precise agent(s) responsible for vasodilation is a subject of wide debate; however, most the recent literature has focused on the likely role of nitric oxide.
Progressive vasodilation leads to the activation of vasoconstrictor and antinatriuretic mechanisms, both in an attempt to restore normal perfusion pressures. Mechanisms involved include the renin-angiotensin system, sympathetic nervous system, and antidiuretic hormone (vasopressin). The ultimate effect is sodium and water retention. In the late stages of cirrhosis, free water accumulation is more pronounced than the sodium retention and leads to a dilutional hyponatremia. This explains why cirrhotic patients with ascites demonstrate urinary sodium retention, increased total body sodium, and dilutional hyponatremia, a challenging concept for many physicians.
The symptoms of ascites vary from patient to patient and largely depend on the quantity of fluid. If trace ascites is present, the patient may be asymptomatic and fluid can be detected only on physical or radiologic examination. If a large amount of fluid is present, the patient might complain of abdominal fullness, early satiety, abdominal pain, or shortness of breath.
Physical examination findings are equally variable. The accuracy of detecting ascites depends on the amount of fluid present and the body habitus of the patient: ascites may be more technically difficult to diagnose in obese patients. If ascites is present, typical findings include generalized abdominal distention, flank fullness, and shifting dullness. If the physical examination is not definitive, abdominal ultrasonography can be used to confirm the presence or absence of ascites.
Two grading systems for ascites have been used in the literature (Table 1). An older system has graded ascites from 1+ to 4+, depending on the detectability of fluid on physical examination. More recently, a different grading system has been proposed, from grade 1 to grade 3. The validity of this grading system has yet to be established.
| Grade | Severity | Score |
|---|---|---|
| 1 | Minimal | 1+ |
| 2 | Moderate | 2+ |
| 3 | Severe | 3+ |
| 4 | Tense | 4+ |
If a noncirrhotic patient develops ascites, diagnostic paracentesis with ascites fluid analysis is an essential part of the medical evaluation. In a patient with well-established cirrhosis, the exact role of a diagnostic paracentesis is less clear. Our opinion is that for a highly functional outpatient with documented cirrhosis, the new development of ascites does not routinely require paracentesis. Cirrhotic patients should, however, undergo paracentesis in the case of unexplained fever, abdominal pain, or encephalopathy or if they are admitted to the hospital for any cause. It is common for hospitalized cirrhotic patients to have infected ascites fluid (spontaneous bacterial peritonitis, SBP) even if no symptoms are present. This is particularly true in the case of a significant gastrointestinal hemorrhage.
Complications from abdominal paracentesis are rare, occurring in less than 1% of cases. A low platelet count or elevated prothrombin time is not considered a contraindication, and prophylactic transfusion of platelets or plasma is almost never indicated. Insertion of the paracentesis needle is most commonly performed in the left or right lower quadrant, but it can also be performed safely in the midline. An abdominal ultrasound can guide the procedure if the fluid is difficult to localize or if initial attempts to obtain fluid are unsuccessful.
Valuable clinical information can often be obtained by gross examination of the ascites fluid (Table 2). Uncomplicated cirrhotic ascites is usually translucent and yellow. If the patient is deeply jaundiced, the fluid might appear brown. Turbidity or cloudiness of the ascites fluid suggests that infection is present and further diagnostic testing should be performed. Pink or bloody fluid is most often caused by mild trauma, with subcutaneous blood contaminating the sample. Bloody ascites is also associated with hepatocellular carcinoma or any malignancy-associated ascites. Milky-appearing fluid usually has an elevated triglyceride concentration. Such fluid, commonly referred to as chylous ascites, can be related to thoracic duct injury or obstruction or lymphoma, but it is often related primarily to cirrhosis.
| Color | Association |
|---|---|
| Translucent or yellow | Normal/sterile |
| Brown | Hyperbilirubinemia (most common) Gallbladder or biliary perforation |
| Cloudy or turbid | Infection |
| Pink or blood tinged | Mild trauma at the site |
| Grossly bloody | Malignancy Abdominal trauma |
| Milky ("chylous") | Cirrhosis Thoracic duct injury Lymphoma |
Many ascites fluid tests are currently available, yet the optimal testing strategy has not been well established. Generally, if uncomplicated cirrhotic ascites is suspected, only a total protein and albumin concentration and a cell count with differential are determined (Box 2). Less than 10mL of fluid is required to perform these basic tests. The albumin concentration is used to confirm the presence of portal hypertension by calculating the serum-to-ascites albumin gradient, or SAAG. The SAAG is determined by subtracting the ascites albumin value from a serum albumin value obtained on the same day:
albuminserum − albuminascites = SAAG
The SAAG has been proved in prospective studies to categorize ascites better than any previous criteria. The presence of a gradient higher than 1.1g/dL indicates that the patient has portal hypertension-related ascites with 97% accuracy. Portal hypertension is usually caused by liver cirrhosis or, less commonly, outflow obstruction from right-sided heart failure or Budd-Chiari syndrome. A SAAG value lower than 1.1g/dL indicates that the patient does not have portal hypertension-related ascites, and another cause of the ascites should be sought. Determination of the SAAG does not need to be repeated after the initial measurement.
| Routine |
|---|
| Cell count with differential |
| Albumin |
| Culture* |
| Sometimes Useful |
| Lactose dehydrogenase level |
| Glucose |
| Amylase |
| Triglyceride |
| Bilirubin |
| Cytology |
| Tuberculosis smear and culture |
| Rarely Helpful |
| pH |
| Lactate |
| Gram stain |
*If infection is suspected and/or corrected polymorphonuclear count is ≥250 cells/mm3.
The cell count and differential are used to determine if the patient is likely to have SBP. Patients with an ascites polymorphonuclear (PMN) count greater than 250 cells/mm3 should receive empiric antibiotics, and additional fluid should be inoculated into blood culture bottles to be sent for culture. The PMN count is calculated by multiplying the white cells/mm3 by the percentage of neutrophils in the differential. In a bloody sample, which contains a high concentration of red blood cells, the PMN count must be corrected: 1 PMN is subtracted from the absolute PMN count for every 250 red cells/mm3 in the sample.
Based on clinical judgment, additional testing can be performed on ascites fluid including total protein, lactate dehydrogenase (LDH), glucose, amylase, triglyceride, bilirubin, cytology, or tuberculosis smear and culture. These tests are generally only useful when there is suspicion of a condition other than sterile cirrhotic ascites. Tests that are not routinely helpful include determination of pH, lactate levels, and Gram staining. Results of Gram staining are of particular low yield unless a large concentration of bacteria, such as in the case of a free gut perforation, is suspected.
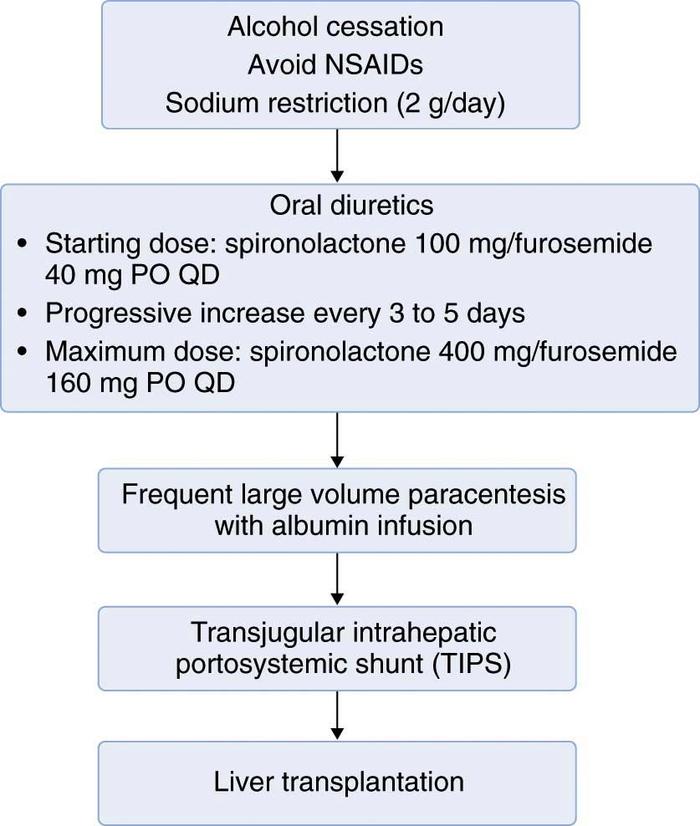
Successful treatment of cirrhotic ascites is defined as the minimization of intraperitoneal fluid without intravascular volume depletion. Despite a lack of data supporting decreased mortality, minimizing the amount of ascites fluid can decrease infection-related morbidity in the cirrhotic patient. Treatment of ascites can dramatically improve quality of life by decreasing abdominal discomfort or dyspnea, or both. General ascites management in all patients should include minimizing consumption of alcohol, nonsteroidal anti-inflammatory drugs (NSAIDs), and dietary sodium. The use of more-aggressive interventions largely depends on the severity of ascites and includes oral diuretics, therapeutic (or large-volume) paracentesis, transjugular intrahepatic portosystemic shunt (TIPS), and orthotopic liver transplantation (Figure 1).
All patients with cirrhotic ascites should be encouraged to minimize consumption of alcohol. Even if alcohol is not the cause of their liver disease, cessation can lead to decreased fluid and improved response to medical therapies. Patients with ascites should also minimize use of all NSAIDs; these agents inhibit the synthesis of renal prostaglandin and can lead to renal vasoconstriction, decreased diuretic response, and acute renal failure. Finally, ascites patients should be counseled to limit their sodium consumption to no more than 2 g/day. Because fluid passively follows sodium, a salt restriction without a fluid restriction is generally all that is required to decrease the amount of ascites. In patients with minimal fluid, the restriction of alcohol, NSAIDs, and salt may be all that is needed to control ascites formation adequately.
Patients with moderate fluid overload who do not respond to more conservative measures should be considered for pharmacologic therapy. A rapid reduction of ascites is often accomplished simply with the addition of low-dose oral diuretics in the outpatient setting.
First-line diuretic therapy for cirrhotic ascites is the combined use of spironolactone (Aldactone) and furosemide (Lasix). Beginning dosages are 100mg of spironolactone and 40mg of furosemide by mouth daily. If weight loss and natriuresis are inadequate, both drugs can be simultaneously increased after 3 to 5 days to 200mg of spironolactone and 80 mg of furosemide. To maintain normal electrolyte balance, the use of the 100 : 40mg ratio of spironolactone to furosemide is generally recommended. Maximum accepted dosages are 400 and 160mg/day of spironolactone and furosemide, respectively.
The response to diuretics should be carefully monitored on the basis of changes in body weight, laboratory tests, and clinical assessment. Patients on diuretics should be weighed daily; the rate of weight loss should not exceed 0.5 kg/day in the absence of edema and should not exceed 1 kg/day when edema is present. Serum potassium, blood urea nitrogen (BUN), and creatinine levels should be serially followed. In the event of marked hyponatremia, hyperkalemia or hypokalemia, renal insufficiency, dehydration, or encephalopathy, diuretics should be reduced or discontinued. Routine measurement of the urinary sodium level is not necessary, but it can be helpful to identify noncompliance with dietary sodium restriction. Patients excreting more than 78 mmol of sodium/day (88 mmol dietary intake – 10 mmol nonurinary excretion) detected on a 24-hour urinary collection should be losing fluid weight. If not, they are noncompliant with their diet and should be referred to a dietician. The spot urine sodium-to-potassium ratio might ultimately replace the cumbersome 24-hour collection: A random urine sodium concentration higher than the potassium concentration has been shown to correlate with a 24-hour sodium excretion higher than 78 mmol/day with approximately 90% accuracy. Because of the potentially severe complications associated with diuretic use, patients with ascites should be assessed by a health care provider at least once weekly until they are clinically stable.
Large-volume ascites is defined as intraperitoneal fluid in an amount that significantly limits the activities of daily life. With additional fluid retention, the abdomen can become progressively distended and painful. This is commonly referred to as massive or tense ascites.
Therapeutic (or large-volume) paracentesis is a well-established therapy for large-volume ascites. However, the use of postprocedural colloid, usually albumin, continues to be a controversial issue. Studies have shown that patients who do not receive intravenous albumin after large-volume paracentesis develop significantly more changes in their serum electrolyte, creatinine, and renin levels. The clinical relevance of these findings, however, is not well established. In fact, no study to date has been able to demonstrate decreased morbidity or mortality in patients given no plasma expanders compared with patients given albumin after paracentesis. In view of the high cost of albumin and its uncertain clinical role, more studies certainly need to be conducted. Until these studies are carried out, current practice guidelines state that it is reasonable, although not mandatory, to give albumin for paracenteses greater than 5 L. Although no direct comparisons have been studied, 25% albumin at doses of 5 to 10 g/L of ascites removed is generally used.
To prevent the reaccumulation of ascites fluid, patients with large-volume ascites should be counseled about limiting consumption of alcohol, NSAIDs, and sodium. They should also be placed on an aggressive diuretic regimen. Diuretic-sensitive patients are generally treated with lifestyle modifications and medications, not serial paracentesis.
Refractory ascites occurs in 5% to 10% of cirrhotic ascites patients and portends a poor prognosis. The definition of refractory ascites is (1) lack of response to high-dose diuretics (400mg of spironolactone and 160mg of furosemide/day) while remaining compliant with a low-sodium diet or (2) frequent ascites recurrence shortly after therapeutic paracentesis. Patients with recurrent side effects from diuretic therapy, including symptomatic hyponatremia, hyperkalemia or hypokalemia, renal insufficiency, or hepatic encephalopathy, are also considered to have refractory ascites. Treatment options include large-volume paracentesis with albumin infusion, placement of a TIPS, or liver transplantation. Surgical shunts (e.g., LeVeen or Denver shunt) have essentially been abandoned because controlled trials have shown poor long-term patency, excessive complications, and no survival advantage over medical therapy.
Frequent therapeutic paracentesis with or without albumin infusion is the most widely accepted treatment for patients with refractory ascites (see “Large-Volume Ascites” for controversy and dosing of albumin use). For those who have loculated fluid or are unwilling or unable to receive frequent paracentesis, TIPS placement can also be considered. In the appropriately selected patient, TIPS is highly effective for preventing ascites recurrence by decreasing the activity of sodium-retaining mechanisms and improving renal function. Ongoing studies will determine whether TIPS might also provide a survival benefit.
In the United States, TIPS is most commonly performed under conscious sedation by an interventional radiologist. The portal system is accessed through the jugular vein, and the operator inserts a self-expanding shunt between the portal (high-pressure) and hepatic (low-pressure) veins. The ultimate goal of the procedure is to lower portal pressures to less than 12mm Hg, the level at which ascites begins to accumulate. Complications are relatively common and include hemorrhage (intrahepatic or intra-abdominal) and stent stenosis or thrombosis. Other important complications include hepatic encephalopathy and decompensation of liver or cardiac function. Therefore, TIPS is generally not recommended for patients with pre-existing encephalopathy, an ejection fraction lower than 55%, or a Child-Pugh Score higher than 12 (Table 3). Additional disadvantages of the procedure are high cost and lack of availability at some medical centers.
| Clinical or Biochemical Parameter | Points |
||
|---|---|---|---|
| 1 | 2 | 3 | |
| Bilirubin (mg/dL) | <2 | 2-3 | >3 |
| Albumin (g/dL) | >3.5 | 2.8-3.5 | <2.8 |
| Ascites | Absent | Moderate | Tense |
| Encephalopathy | Absent | Moderate (I or II) | Severe (III or IV) |
| Prothrombin time | |||
| Seconds prolonged or | <4 | 4-6 | >6 |
| International normalized ratio (INR) | <1.7 | 1.7-2.3 | >2.3 |
*Child-Pugh score: A = 5-6, B = 7-9, C = 10-15.
Liver transplantation is the ultimate treatment for cirrhosis and cirrhotic ascites. Appropriate timing for referral is debated, but should be considered when a cirrhotic patient first presents with a complication from cirrhosis, such as ascites. Because refractory ascites portends a particularly poor prognosis, immediate referral to an experienced liver transplantation center is recommended.
The 2-year survival rate for a patient with cirrhotic ascites is approximately 50%. Once a patient becomes refractory to routine medical therapy, 50% die within 6 months and 75% within 1 year. Because liver transplantation is associated with 2-year survival rates of almost 85%, it should be considered as an important treatment option in all appropriate patients.
Many with ascites will develop infection, most often without a known precipitating factor (such as diverticulitis, bowel perforation, etc). This is referred to as spontaneous bacterial peritonitis (SBP). SBP should be suspected whenever there is clinical deterioration in a cirrhotic with ascites. Diagnosis rests on ascitic fluid cell count (more than 250 polymorphonuclear cells/mm3) or a positive ascitic fluid culture. Treatment should be undertaken whenever SBP is suspected. Most often intravenous therapy with a third undertaken whenever SBP is suspected. Most often intravenous therapy with a third generation cephalosporin (e.g., cefotaxime 2gm every 8 hours) is used. Quinolones may also be effective, including oral agents such as ofloxacin (400mg twice per day). Intravenous albumin (1.5gm/kg body weight on day zero and 1.0gm/kg on day 3 has been shown to improve survival in SBP, particularly in those with renal insufficiency and should be used if the creatinine or BUN are elevated. Antibiotic treatment should be continued for 5 days. The presence of bacteremia does not influence treatment duration. Longer therapy may be warranted in individual cases. Those who survive SBP are at risk for a second bout. Prophylactic antibiotics are recommended, e.g., norfloxacin 400mg daily, or trimethoprim/sulfamethoxazole (160/800). A clinical practice guideline updated in 2009 suggests a role for SBP prophylaxis even in those who have never had SBP. Primary SBP prophylaxis should be considered in patients with ascites containing <1.5 m/dl and one or more of the following: serum creatinine >1.2mg/dl; BUN >25mg/dl; serum sodium concentration <130mEq/L, Childs Pugh score >9 together with a bilirubin >3mg/dl.
A feared complication of advanced liver disease is hepatorenal syndrome (HRS). In its most virulent form (type I) there is an inexorable worsening of renal function reflecting in rising creatinine and BUN, resulting in death. This syndrome almost always occurs in the setting of ascites. A diagnosis is established when other causes of acute renal insufficiency are excluded, especially hypovolemia due either to diuretic use, infection, or bleeding. HRS is established in a patient with a creatinine of >1.5mg/dl that does not improve with withdrawal of diuretics, volume expansion with albumin, absence of obstructive or parenchymal renal disease (indicated by ultrasonography, proteinuria), and absence of recent use of nephrotoxic drugs or administration of IV contrast material. Treatment of HRS is frustrating and often unsuccessful. Studies suggest a possible role for use of intravenous albumin (e.g., 25 grams daily) together with octreotide (200ug sc TID) and midodrine (5mg TID, titrating to a maximum of 12.5mg TID). Referral for liver transplantation services should be considered for appropriate patients. A less severe form of HRS (type II) is recognized. In this variant, non-progressive renal impairment without another etiology than cirrhosis is seen.