Published: August 2010
Liver disease related to hepatitis B remains an important public health concern and a major cause of morbidity and mortality. It also presents a common challenging problem for practicing physicians.
Hepatitis B is found throughout the world, but its prevalence varies greatly; it is especially high in Asia, sub-Saharan Africa, and the South Pacific, as well as in specific populations in South America, the Middle East, and the Arctic.1 Prevalence in the United States varies, based on the population makeup, including the extent of the immigrant population from endemic areas, and on risk factors and behavior, such as the prevalence of intravenous drug use and homosexual practices. Public health agencies estimate that there are about 1.25 million people infected in the United States, but 2 billion people infected worldwide, with approximately 5% of the world's population (or 350 million people) being carriers of chronic hepatitis B.2 In a typical year, 70,000 Americans become infected with chronic hepatitis B virus (HBV), and approximately 5000 patients with chronic hepatitis B die of complications caused by the disease. Worldwide, chronic hepatitis B is the tenth leading cause of death.
Hepatitis B was first discovered in 1963 by Dr. Baruch Blumberg and colleagues, who identified a protein (the “Australia antigen” that reacted to antibodies from patients with hemophilia and leukemia. The association of this protein with infectious hepatitis was discovered 3 years later by several investigators, and the virus was specifically seen by electron microscopy in 1970.3
HBV is a double-stranded hepatotropic DNA virus belonging to the family Hepadnaviridae. The virus infects only humans and some other nonhuman primates. Viral replication takes place predominantly in hepatocytes and, to a lesser extent in the kidneys, pancreas, bone marrow, and spleen. The viral genome is 3.2 kb in length and possesses four partially overlapping open-reading frames that encode various antigens.4 The intact virion is a spherical double-shelled particle with an envelope of hepatitis B surface antigen (HBsAg), an inner nucleocapsid of core antigen (HBcAg), and an active polymerase enzyme linked to a single molecule of double-stranded HBV DNA. Significant variability of the nucleotide sequence exists, and the virus can be subdivided into eight different genotypes, based on the degree of variation. The clinical importance of these is still uncertain, however.
Although HBV can survive outside the body for up to 1 week—and therefore, might be transmitted via indirect contact, such as from open sores—hepatitis B is spread predominantly parenterally, through intimate personal contact, and perinatally. Persons at risk include intravenous drug users, children of mothers with HBV, men who have sex with men, patients on hemodialysis, and those exposed to blood or blood products.
The incubation period of HBV ranges from 45 to 160 days (mean, 100 days). The acute illness is usually mild, particularly in children. In adults, as many as 30% to 50% present with jaundice, and hepatitis may be fulminant in 0.1% to 0.5% of those with acute hepatitis B infection. Symptoms therefore range widely in severity, from asymptomatic subclinical infection to fulminant fatal disease. An insidious onset of nausea, anorexia, malaise, and fatigue, or flulike symptoms, such as pharyngitis, cough, coryza, photophobia, headache, and myalgias, can precede the onset of jaundice. Fever is uncommon, unlike with hepatitis A infection. These symptoms abate with the onset of jaundice, although anorexia, malaise, and weakness can persist. Physical examination features are nonspecific but can include mild enlargement and slight tenderness of the liver, mild splenomegaly, and posterior cervical lymphadenopathy in 15% to 20% of patients. Fulminant disease (acute liver failure) manifests with a change in mental status (encephalopathy) and coagulopathy.5
The risk of developing chronic infection, or the carrier state, defined as the persistence of HBsAg in the blood for longer than 6 months, depends on the age and immune function of the patient at the time of initial infection. Ninety percent of infected newborns, 30% of children younger than 5 years, and 10% of adults progress to chronic infection. Of these carriers, 15% to 40% develop hepatitis B-related sequelae in their lifetimes. Patients with chronic infection spontaneously clear surface antigen at a rate of 0.5% per year.6 Patients with chronic hepatitis B can develop extrahepatic manifestations, including arthralgias, mucocutaneous vasculitis, glomerulonephritis, and polyarteritis nodosa. The glomerulonephritis of hepatitis B occurs more commonly in children than in adults and is usually characterized by the nephrotic syndrome, with little decrease in renal function. Polyarteritis nodosa occurs primarily in adults and is marked by a sudden and severe onset of hypertension, renal disease, and systemic vasculitis with arteritis in the vessels of the kidneys, gallbladder, intestine, or brain. Other rare extrahepatic manifestations are mixed essential cryoglobulinemia, pericarditis, and pancreatitis.
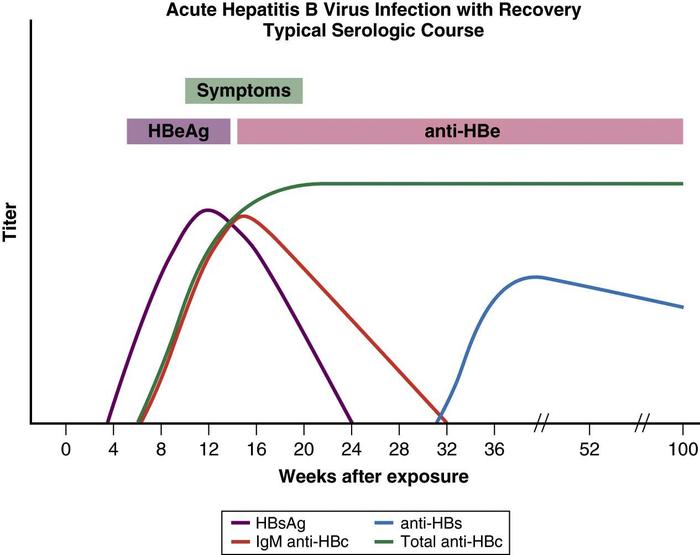
Viral and immune markers are detectable in blood, and characteristic antigen-antibody patterns evolve over time. The first detectable viral marker is HBsAg, followed by hepatitis B e antigen (HBeAg) and HBV DNA. Titers may be high during the incubation period, but HBV DNA and HBeAg levels begin to fall at the onset of illness and may be undetectable at the time of peak clinical illness.7 Core antigen does not appear in blood, but antibody to this antigen (anti-HBc) is detectable with the onset of clinical symptoms.
The IgM fraction is used in an important diagnostic assay for acute hepatitis B infection. Before current molecular assays were available, it was the only marker detectable in the window period, the time between the disappearance of HBsAg and the appearance of anti-HBs. Patients who clear the virus lose HBsAg and develop anti-HBsAb, a long-lasting antibody associated with immunity. The presence of anti-HBsAb and anti-HBcAb (IgG) indicates recovery and immunity in a previously infected person, whereas a successful vaccination response produces antibody only to HBsAg (Box 1).
| Box 1: Serologic Patterns for Hepatitis B |
|---|
| Immunity |
| Natural Exposure |
|
| Vaccination |
|
| Acute Infection |
|
| Chronic Infection |
|
HBcAb, hepatitis B core antibody; HBsAb, hepatitis B surface antibody; HBeAg, hepatitis B e antigen; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus; Ig, immunoglobulin.
© 2005 The Cleveland Clinic Foundation.
HBeAg is another viral marker detectable in blood. It correlates with active viral replication and therefore high viral load and infectivity. The antigen is synthesized from a strand of DNA immediately preceding the area that codes for the core antigen.8 A mutation in this area can occur, preventing the production of the HBeAg. Such viruses are present throughout the world, particularly in Asia and the Mediterranean, and are known as precore mutants. The presence of a precore or core mutant, causing HBeAg-negative chronic hepatitis, typically implies disease of longer standing and therefore a higher risk of cirrhosis.
The hepatitis B virus is not cytopathic, and liver injury in chronic hepatitis B is believed to be immunologically mediated. Thus, the severity and course of disease do not correlate well with the level of virus in serum or the amount of antigen expressed in the liver. Antigen-specific cytotoxic T cells are believed to play a role in the cell injury in hepatitis B, but they ultimately account for viral clearance. Specific cytokines produced by cytotoxic and other T cells also have antiviral effects, contributing to viral clearance without cell death. The lack of a vigorous and specific CD8+ cytotoxic T cell and CD4+ helper T cell response can allow chronic infection to develop. Recruitment of nonspecific T cells then results in low-level chronic inflammation and liver damage. Similarly, spontaneous seroconversion from HBeAg to anti-HBeAb during chronic hepatitis B is also immunologically mediated, as is suggested from the transient flare of disease that often immediately precedes clearance of HBeAg.7
Acute hepatitis B is diagnosed by detecting HBsAg and IgM core antibody, or core antibody alone, in the window period. IgM core antibodies are lost within 6 to 12 months of the onset of illness. Biochemically, serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels can increase to between 500 to 5000 U/L and fall after the acute phase of infection. Serum bilirubin levels seldom increase above 10 mg/dL, the alkaline phosphatase level and prothrombin time are usually normal or mildly elevated (e.g., 1 to 3 seconds), and the serum albumin level is normal or minimally depressed. Peripheral blood counts may show mild leukopenia, with or without relative lymphocytosis. Loss of HBsAg and the development of HBsAb signify recovery from the acute infection and the development of immunity (Fig. 1).
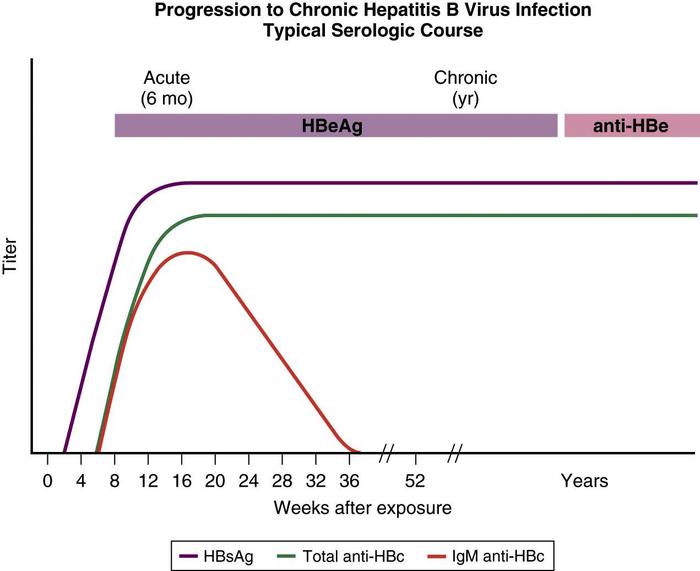
Chronic hepatitis B is defined as the persistence of HBsAg in serum for at least 6 months. Patients with chronic infection may be divided into those with evidence of active replication, typically associated with abnormal transaminase levels and higher viral loads, and those in the nonreplicative state, associated with decreased markers of liver inflammation and damage and lower viral loads. Transaminase levels may be normal, or they may be increased anywhere from 1 to 10 times the upper limit of normal. Levels of HBV DNA are usually in the range of 105 genome copies/mL, which are readily detectable by hybridization techniques, but the absolute level can fluctuate.
HBeAg in serum reflects active viral replication, and the clinical outcome of infection is correlated with HBeAg status. Conversion to HBeAg-negative and HBeAb-positive status in patients with chronic hepatitis B typically leads to decreased inflammation, with normalizing transaminase levels and decreased levels of HBV DNA in serum: the inactive carrier state. The e antigen marker is also absent in patients with core or precore mutants. Using conventional hybridization assays, HbsAg carriers do not have detectable HBV DNA in serum. Testing for HBV DNA with more sensitive techniques, such as the polymerase chain reaction (PCR) assay, however, usually demonstrates low levels of viral DNA in serum in these carriers (Fig. 2).
The course of chronic hepatitis B is variable. Spontaneous loss of HBeAg occurs at a rate of 8% to 12% per year, associated with a decrease in HBV DNA below levels detected by hybridization techniques. Loss of HBsAg occurs less often (<1%/year). Chronically infected patients without active liver disease or viral replication (inactive carriers) generally have a benign course, with a smaller likelihood of progressing to cirrhosis. Patients who continue to have active viral replication with high levels of HBV DNA and HBeAg in serum have progressive liver injury, and cirrhosis and end-stage liver disease can develop. A transient flare of disease often precedes remission. Loss of HBeAg is not always followed by permanent resolution of disease and disease flares can occur, particularly if a patient is treated with steroids or other immunosuppressive medications. Patients who revert to chronic HBeAg-positive status tend to develop cirrhosis at a substantially increased rate compared with those who remain HBeAg-negative.9 Patients infected with a core or precore mutant strain, who continue to have high DNA levels and evidence of ongoing hepatic inflammation, tend to have a higher risk of disease progression than patients who are HBeAg-positive.
Chronic HBV infection is associated with a ten-fold increase in the risk of developing hepatocellular carcinoma (HCC). This risk is further magnified in the setting of ongoing inflammation: In patients with both HBsAg and HBeAg, the risk increases to 60-fold compared with the general population.10 Older men with cirrhosis and those coinfected with hepatitis C are at greatest risk. In regions where HBV is endemic, HCC is the leading cause of cancer-related death. It is therefore recommended that HBV carriers, particularly those at highest risk (men older than 45 years, patients with cirrhosis, and those with a family history of liver cancer) should be screened with ultrasound and alpha-fetoprotein testing for HCC at 6-month intervals.11
Effective vaccines for HBV, defined as inducing better than 90% protection against HBV, have been available in the United States since 1982. Hepatitis B vaccine has been described as the first effective anticancer vaccine, and its use has been promoted by the World Health Organization as routine care worldwide since 1997. Early strategies targeted high-risk groups, but they were not successful in materially decreasing incidence rates. Therefore, universal vaccination for HBV has been recommended for infants by the American Academy of Pediatrics since 1991. For patients with a documented exposure, postexposure prophylaxis consists of a single dose of hepatitis B immunoglobulin (HBIg) injected intramuscularly, followed immediately by HBV vaccination. Two recombinant hepatitis B vaccines are available in the United States, Engerix-B. and Recombivax HB. For adults, the recommended regimen is three injections (20 µg of Engerix-B or 10 µg of Recombivax HB) intramuscularly in the deltoid muscle at 0, 1, and 6 months. The seroconversion rate is greater than 90% in adults but may be lower in certain persons, depending on comorbid diseases or genetic factors, as well as in smokers, the obese, older adults, or patients who are immunocompromised. These patients might require higher doses and more injections.
Prevaccination screening for anti-HBs is not recommended except for adult patients who are likely to have been previously exposed, including those in high-risk groups (e.g., injection drug users, male homosexuals). Postvaccination testing for anti-HBs to document seroconversion is not routinely recommended, except for persons who are at risk for lack of response or continued exposure. Booster doses may be appropriate for high-risk patients if titers of anti-HBs fall below what is considered protective (10 IU/mL). The vaccine should be routinely administered to everyone younger than 18 years and to adults at risk of exposure. It should be given to neonates of HBV-infected mothers together with HBIg.12
In acute hepatitis B, treatment is supportive. Although several case series have been published, there is no clear evidence that early therapy with antiviral agents for acute hepatitis B decreases the risk of chronicity or speeds recovery. Most patients with acute icteric hepatitis B recover without residual injury or chronic hepatitis. Patients should be followed with repeat testing for HBsAg and ALT levels to determine whether seroconversion and clearance of surface antigen have occurred.13
In chronic hepatitis B, therapy is administered to suppress viral replication and prevent progression of liver disease. Although several end points are therefore important, the ability of any medication to prevent liver damage may be related to specific targets, including the prevention of inflammation (leading to decreased liver enzyme levels, a biochemical end point), or the ability of a drug to induce seroconversion (from HBeAg-positive to HBeAg-negative) or a change in fibrosis (i.e., a decrease in scar tissue on repeat liver biopsy). Because the likelihood of developing anti-HBs, and therefore recovery with long-term protection from hepatitis B, is fairly low, measured outcomes of treatment focus on rates of normalization of liver enzyme levels, decreases in viral DNA levels, or seroconversion—that is, from HBeAg-positive to HBeAg-negative, with a positive HBeAb.
In the absence of cirrhosis, therapy is not routinely recommended for patients with normal enzyme levels whether they are chronic inactive carriers or based on their HBeAg status.14 Therapy is recommended for patients with evidence of active damage to the liver, such as those with abnormal transaminase levels (an ALT level more than twice the upper limit of normal). A liver biopsy before therapy is the gold standard to assess the degree of necroinflammatory activity and fibrosis. Although the data are still evolving, the most recent recommendations of the American Association for the Study of Liver Diseases (AASLD) also include treatment of patients with compensated and decompensated cirrhosis and measurable HBV DNA (>2000 IU/ml) regardless of HBeAg status or degree of elevation of ALT level.14 This approach is supported by several studies that have shown a decreased rate of development of progressive liver disease or complications in treated patients.
Six agents have been approved by the U.S. Food and Drug Administration (FDA) to treat hepatitis B. Interferon alfa, available since 1992 and injected subcutaneously at a dosage of 5 MU daily, has direct antiviral activity as well as effects on the host immune system. The major side effects of interferon include fatigue, muscle aches, fever, depression, and irritability. Uncommon severe side effects include exacerbation of depression, psychosis, renal and cardiac failure, bacterial infections, and induction of autoimmunity. The FDA approved the use of long-acting interferon (peginterferon alfa-2a, at a dose of 180 µg for 48 weeks) in 2005 for treating patients with chronic hepatitis B; the side-effect profile of peginterferon alfa-2a is very similar to that of shorter-acting interferon. Other treatments available are oral agents and include nucleoside or nucleotide analogues, which interfere with the replication of the hepatitis B virus. The advantages of these medications include a relatively more benign side effect profile compared with interferon; however, the durability of response after treatment might not be as reliable as that of interferon. The first of these was lamivudine, approved by the FDA in 1998. Other medications available for treating HBV include adefovir, approved by the FDA in September 2002, entecavir, approved in March 2005, and telbivudine, approved in October 2006.
Patients who are HBeAg-positive and have evidence of liver disease should be treated. The choice among treatment options is dictated by considerations of the likelihood of response, cost, length of treatment, and side-effect profile, as well as the likelihood of developing resistance. There are some data regarding the likelihood of treatment response in patients treated with interferon, with a higher chance of success in patients with high ALT levels but low HBV DNA levels. Analogously, lamivudine is more likely to be effective in patients with increased ALT levels or inflammation on liver biopsy. Comparable predictors of response for the other antivirals have not been established.
The response rate for these different therapies in this population, defined as seroconversion (from HBeAg-positive to HBeAg-negative, with a positive HBeAb) is variable; published rates are 12% (with adefovir), 16% to 18% with lamivudine, 21% with entecavir, 26% with telbivudine, and 32% to 33% with peginterferon alfa-2a or interferon. Other end points (normalization of liver enzyme levels or improvement in liver histology) are typically seen in 50% to 70% of treated patients. Patients with a beneficial response to interferon therapy often develop a flare of disease, with elevations of serum ALT to levels two to three times the baseline before normalization occurs. Because of the possibility that a flare of liver disease can lead to decompensation, the use of interferon in cirrhotic patients is not recommended. Disease flares, by comparison, are not typically seen in patients treated with lamivudine or adefovir. Preliminary data have suggested that entecavir might also be safe in cirrhotic patients.
Treatment for patients with HBeAg-negative disease is also possible. Several studies have shown efficacy with each of the various approved therapies, in terms of loss of hepatitis B viral DNA or normalization of liver enzyme levels (in approximately 60%-70%). Unfortunately, the response rates are often not sustained, with very high relapse rates after therapy is stopped. As a result, the optimal duration of therapy is not defined in this population.
An important consideration in patients treated with any of the nucleoside or nucleotide analogues is the possibility of emergence of resistant mutants, which increases with increasing duration of treatment. This is particularly true with lamivudine treatment, for which rates of resistance range from 24% at 1 year to 42% by year 2 of continued therapy. Lamivudine resistance is manifested by the reappearance of HBV DNA in serum, most commonly with the YMDD mutant, characterized by an amino acid substitution in the HBV DNA polymerase. The outcomes in these patients are variable, but the emergence of a mutant virus can lead to a serious flare of liver disease. Patients therefore should be monitored for the development of resistance and considered for treatment with another antiviral. Other antivirals are associated with a much lower rate of resistance, but none of them is immune from this possibility. Combination therapy with several agents is likely more effective in preventing the development of resistance, but optimal combinations to improve response rates and clinical outcomes have not yet been defined.
Thus, although the introduction of nucleotide or nucleoside analogues represents a significant advance in the management of chronic hepatitis B, many questions remain regarding optimal dosing, duration, and possible combinations to prevent resistance, increase long-term suppression, or promote eventual clearance. A number of other drugs, including emtricitabine, clevudine, famciclovir, and tenofovir, have also shown some efficacy, often in patients coinfected with HIV, and are therefore being further studied in a number of clinical trials. These emerging therapies, including newer and more potent antiviral agents, coupled with aggressive worldwide vaccination policies, lend promise to the hope that hepatitis B will one day be controlled.