Published: April 2018
Expire: April 2021
Multiple Sclerosis (MS) is a chronic inflammatory, demyelinating, and neurodegenerative disorder of the central nervous system (CNS) that affects the white and grey matter of the brain, spinal cord, and optic nerve. MS is one of the most common causes of non-traumatic disability among young and middle-aged adults. Direct MS-related healthcare costs are estimated to be more than $10 billion annually in the United States.1 As symptoms of MS are extremely variable and often quite subtle, diagnosis and management have been greatly enhanced by the use of magnetic resonance imaging (MRI). Therapies that target inflammation and slow progression of disease are available; therefore, early diagnosis and treatment are important in limiting the impact of this potentially devastating disease. Complementary approaches such as symptom management and healthy lifestyle practices also have an important role in MS care.
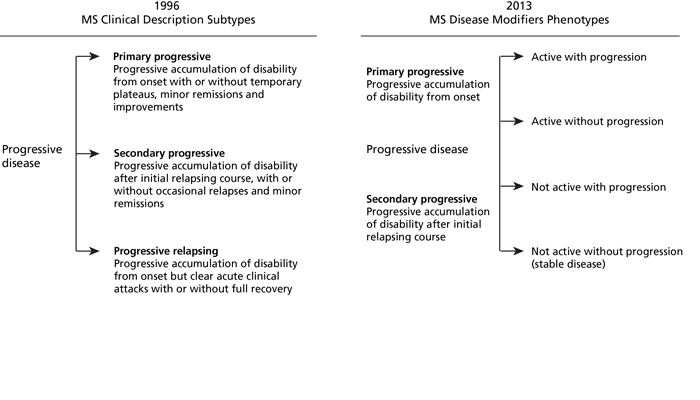
There are several different forms of MS. Since these classifications were based upon clinical characteristics, they are empiric and do not reflect specific biologic pathophysiology. Nonetheless, they provide an organized framework for diagnosis and long-term management. Approximately 85% of patients present with a relapsing-remitting MS (RRMS) disease course at onset, where symptoms appear for several days to weeks, after which they usually resolve spontaneously.2 After tissue damage accumulates over many years and reaches a critical threshold, about two-thirds of the patients transition to secondary progressive MS (SPMS), where pre-existing neurologic deficits gradually worsen over time. Relapses can be seen during the early stages of SPMS, but are uncommon as the disease further progresses. About 10% to 15% of patients have gradually worsening manifestations from the onset without clinical relapses, known as primary progressive MS (PPMS).3 Patients with PPMS tend to be older, have fewer abnormalities on brain MRI, and generally respond less effectively to standard MS therapies.4
MS affects approximately 1 million individuals in the US and 2.5 million worldwide.5,6 Initial symptoms typically occur between 20 and 50 years of age, and women have about 3 times increased likelihood of developing MS compared with men.1 Although MS is more frequently seen in white than African American and Hispanic populations, the latter groups overall have poorer disease outcomes in that they accumulate disability more quickly, suggesting more destructive tissue injury in these groups. The prevalence of MS varies by location and generally increases the further one travels from the equator in either hemisphere. It remains unclear whether this altered incidence represents an environmental influence (eg, vitamin D deficiency), genetic difference, variable surveillance, or other, as yet unidentified, differences.
Early in the disease course, MS involves recurrent bouts of CNS inflammation that results in damage to both the myelin sheath surrounding axons as well as the axons themselves. Histologic examination reveals foci of severe demyelination, decreased axonal and oligodendrocyte numbers, and glial scarring. The exact cause of inflammation remains unclear, but an autoimmune response directed against CNS antigens is suspected.
In progressive MS, inflammation is a less defining pathological hallmark. Instead, progressive MS is characterized by neurodegeneration of the white and grey matter resulting in brain and spinal cord atrophy on a background of mild-moderate inflammation.7 Predominant factors driving neurodegeneration include mitochondrial dysfunction due to defective oxidative phosphorylation and nitric oxide production, resulting in a chronic state of virtual hypoxia due to unmet energy demands,8 and age-dependent iron accumulation in myelin and oligodendrocytes leading to oxidative tissue damage.9 Further research is needed to understand how these different pathologic subtypes affect prognosis and response to treatments. Currently, brain biopsy is the only method to definitively determine pathologic subtypes, but studies are underway to find blood, cerebrospinal fluid, and MRI biomarkers.
Historically, MS was classified as an inflammatory disease targeting white matter, with diagnostics and therapeutics focused on this mechanism of pathology. However, more recent imaging and histopathological studies suggest that cortical demyelination plays a crucial role in MS pathogenesis and cognitive dysfunction. Cortical demyelination is now recognized in early MS.10 Although some investigative MRI modalities capture some cortical involvement, including double inversion recovery sequences at 3 tesla and ultra-high field MRI, conventional MRI metrics used in clinical practice do not show these changes well. Likewise, extensive cortical demyelination that is seen in histopathological studies is not clearly demonstrated on any current MRI modality. This pathology/imaging discordance demonstrates that we are still technologically disadvantaged in accurately assessing cortical lesion pathology in the live patient.
In the past, inflammation was thought to involve only demyelination, but pathologic studies have found significant axonal pathology as well. In actively demyelinating MS lesions, an average of more than 11,000 transected axons/mm3 were observed, while control brain tissue had less than one transected axon/mm.11 Significant axonal injury is also observed in cortical demyelinating lesions. Clearly, axonal injury is significant in the early stages of disease.
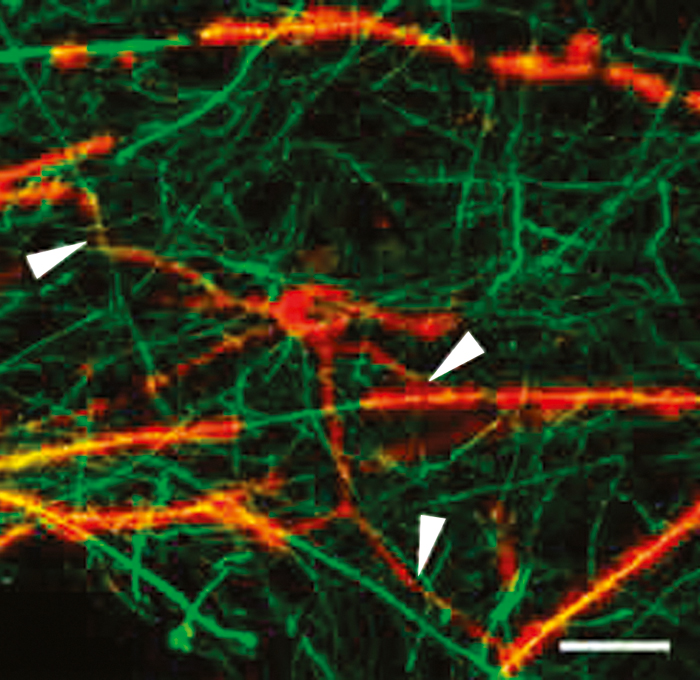
Later in the disease course, gradual progression of disability is observed. However, there is significantly less active inflammation during this period, so clinical progression may arise instead from degenerative changes. Nonetheless, oligodendrocyte progenitor cells capable of remyelinating axons have been observed even in white matter plaques from patients with chronic MS (Figure 1).12 This observation suggests that the potential for remyelination persists even very late in the disease course, which is an encouraging indicator for possible therapeutic targets at this late stage of disease.
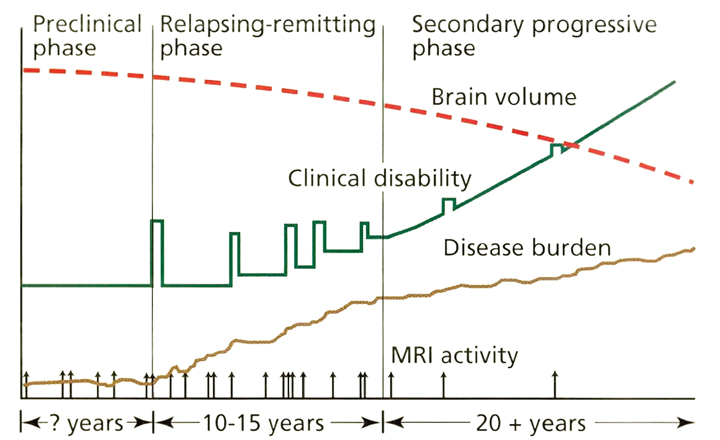
Current concepts of the pathophysiology of MS are illustrated in Figure 2.13 In the preclinical phase, patients may develop lesions characteristic of MS visible on MRI before they phenotypically manifest symptoms, known as radiologically isolated syndrome. In a different scenario, patients may develop MS symptoms compatible with inflammatory demyelination without other characteristic lesions on MRI. This phenomenon is called clinically isolated syndrome.
On average, patients with RRMS experience clinical relapses every 1 to 2 years. Serial MRI studies show that lesions develop up to 10 to 20 times more frequently than clinical relapses Thus, although RRMS appears to have clinically active and quiescent periods, inflammatory lesions are developing and evolving almost continuously. A current hypothesis states that overt progression of disability, which marks the transition from RRMS to SPMS, occurs when ongoing irreversible tissue injury exceeds a critical threshold beyond which the nervous system can no longer compensate. It is thought that at this point the disease has primarily transitioned to a neurodegenerative condition with neurologic deterioration independent of ongoing inflammation, although superimposed inflammation can continue to cause additional injury. An important implication of this hypothesis is that the accumulation of irreversible tissue damage limits the potential for anti-inflammatory disease modifying therapies (DMTs) when used in the progressive stage of the disease. To be maximally effective, DMTs should be started early in patients with RRMS before permanent disability develops. Overall, an incomplete understanding of progressive MS pathogenesis has slowed the development of effective therapies and requires further inquiry.
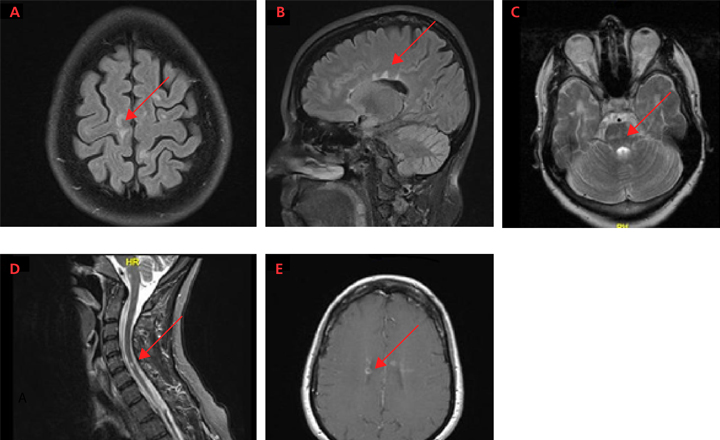
MS can cause a wide variety of neurologic symptoms since it can affect numerous areas of the brain, optic nerve, and spinal cord (Figure 3). Characteristic lesions are located in the periventricular and juxtacortical regions, in addition to the brainstem, cerebellum, spinal cord, and optic nerve. Disease localized to the spinal cord may cause partial or complete transverse myelitis, involving sensory or motor changes involving 1 or both sides of the body. Lhermitte’s phenomenon is a nonspecific symptom whereby flexion of the neck causes an electrical-like shooting sensation that extends into the arms or down the back. It is thought to arise from partially demyelinated tissue, whereby mechanical stimulation leads to axonal activation. Other common symptoms of MS often stemming from spinal cord lesions include bladder and bowel dysfunction. Posterior fossa (eg, brainstem and cerebellum) involvement may present as diplopia, dysphagia, altered sensation or weakness of the face, or ataxia. Inflammation of the optic nerve (optic neuritis) usually presents as blurry vision with painful eye movements, and is often an early clinical manifestation of RRMS.
Of all the lesions in MS, cerebral lesions are the most common but cause the fewest symptoms early in MS. Most cerebral lesions are not located in eloquent regions and so are thus clinically silent and identified only by brain MRI. Very large cerebral lesions may present with weakness or numbness and rarely may cause aphasia or other cortical dysfunction. Cerebral and cortical lesions may also cause subtle symptoms, such as cognitive impairment, fatigue, and affective disorders like depression. Although these symptoms are not uncommon in patients with MS, they are also nonspecific and can be seen in a multitude of disorders.
Symptoms of a clinical relapse typically arise over hours to days, worsen over several weeks, and then gradually subside over several weeks or months. Residual enduring neurologic symptoms are common. The gradual progression of progressive MS can manifest as worsening myelopathy causing asymmetric limb weakness, ataxia, spasticity, and bladder/bowel and sexual dysfunction; impaired mobility; impaired motor dexterity; and cognitive impairment.
There are no pathognomonic clinical, laboratory, or imaging findings in MS. The diagnosis ultimately is a clinical decision based on weighing the factors that support the diagnosis against those that fail to support it or point to the possibility of an alternative diagnosis.
The Schumacher criteria from 1965 capture the essence of the diagnosis of MS: CNS lesions disseminated in space and time and the elimination of alternative diagnoses.14 These core diagnostic characteristics remain relevant today.
The International Panel on MS Diagnosis criteria, also called the McDonald criteria, are diagnostic criteria for MS that incorporate the clinical characteristics and MRI features.15 Revisions were made in 2005, 2010, and most recently in 2017 as a reflection of an increased understanding of the natural history of MS and improved MRI techniques.
The latest version of the McDonald criteria (2017) simplifies the diagnostic process and allows earlier diagnosis (Table 1).16 A diagnosis of clinically definite multiple sclerosis requires fulfillment of dissemination in space and time. Dissemination in space is defined as 1 or more T2-hyperintense lesions in more than 1 characteristic location of MS, which includes the periventricular, juxtacortical, and infrantentorial regions (eg, brainstem and cerebellum), and spinal cord. Cerebrospinal-fluid restricted oligoclonal bands can be used as paraclinical support for an early diagnosis of MS (see Table 1). The 2016 magnetic resonance imaging in multiple sclerosis (MAGNIMS) criteria now include the optic nerve as 1 of the characteristic locations fulfilling an MS diagnosis,17 though it still remains separate from the McDonald criteria. Dissemination in time is defined as 1) the simultaneous presence of a gadolinium-enhancing (GdE) and non-enhancing lesion at any time on initial MRI or 2) a new T2-hyperintense or GdE lesion on follow-up MRI with reference to a baseline scan, irrespective of the timing of the baseline MRI.
| Clinical presentation | Additional findings needed for MS diagnosis |
|---|---|
| ≥ 2 clinical attacks and objective clinical evidence of ≥ 2 lesions |
None; however, magnetic resonance imaging (MRI) is typically obtained to both exclude other diagnoses and stage the severity of disease. |
| ≥ 2 clinical attacks and objective clinical evidence of 1 lesion |
Dissemination in space (DIS)a,b: an additional clinical attack implicating a different central nervous system site OR by MRI |
| 1 clinical attack and objective clinical evidence of ≥ 2 lesions |
Dissemination in time (DIT)a,c: an additional clinical attack OR by MRI OR cerebrospinal fluid-specific oligoclonal bands |
| 1 clinical attack and objective clinical evidence of 1 lesion (MS at first episode if initial MRI criteria are met for DIS and DIT or DIS plus cerebrospinal fluid-specific oligoclonal bands)d | DIS: an additional clinical attack implicating a different CNS cite OR by MRI DIT: an additional clinical attack OR by MRI OR cerebrospinal fluid-specific oligoclonal bands |
aDIS and DIT can be made by clinical features alone or by a combination of clinical and MRI features.
bDIS principle requires that there are asymptomatic lesions typical of MS present in 2 or more sites within the central nervous system: periventricular, subcortical, infrantentorial, and spinal cord.
cDIT principle requires that 2 attacks separated by more than 30 days have occurred in different parts of the central nervous system. MRI criteria for DIT stipulate either an asymptomatic enhancing T2 lesion along with a non-enhancing T2 lesion on any one scan, or a new T2 or gadolinium-enhancing lesion on a follow-up scan.
dIn a patient with a typical clinically isolated syndrome and fulfillment of clinical or MRI criteria for DIS with no better explanation for the clinical presentation, demonstration of cerebrospinal fluid-specific oligoclonal bands allows an MS diagnosis to be made (change from the 2010 McDonald Criteria).
Adapted from AJ Thompson, et al.16
In all cases, the practitioner must rule out better explanations for the clinical presentation other than multiple sclerosis. In the context of the MacDonald criteria, a single episode of demyelination and certain findings on a single MRI can fulfill the diagnostic criteria for MS, even before a second clinical episode or new MRI lesion. The revisions also preserve diagnostic sensitivity and specificity and address their applicability across different populations, allowing for more uniform and widespread use across groups.
In 2013, an international panel of MS experts proposed changes to the classification of MS to more effectively characterize the disease course.18 One of the changes included the categorization of disease as either manifesting active inflammation (‘active’) or no active inflammation (‘non-active’) based on new clinical relapses or new T2 or GdE MRI lesions or in combination within the past year. Another change was the categorization of disease based on the presence or absence of continued gradual clinical decline (with progression or without progression) (Figure 4).18 These disease classifications were intended to provide a clearer conceptualization of progressive MS and its differentiation from active inflammation.
Although the diagnosis of MS cannot be based on MRI alone, typical MRI lesions in the periventricular and juxtacortical regions, as well as the brainstem, cerebellum, and spinal cord can raise the suspicion of MS, warranting further diagnostic workup or monitoring. MRI is typically obtained at the time of diagnosis to both exclude other diagnoses and stage the severity of disease. Patients with a typical history of MS without typical MRI findings are highly unusual and should prompt consideration of an alternative diagnosis.
Management of MS requires multiple therapeutic approaches. The current goals of MS management involve the treatment of acute relapses, prevention of new disease activity and disability progression, management of symptoms that affect quality of life, and adherence to a healthy lifestyle.
Several studies have found that treatment with corticosteroids can shorten the length of relapses and may even improve long-term outcomes.19,20 A typical regimen is 500 mg to 1,000 mg of intravenous methylprednisolone with or without a tapering dose of oral prednisone over several weeks. The standard protocol at the Cleveland Clinic is intravenous methylprednisolone 1,000 mg daily for 3 to 5 days, followed by a 12-day prednisone taper (60 mg daily, decreasing by 20 mg every 4 days). Evaluation of a relapse should include a search for precipitating factors such as fever, upper respiratory illness, or bladder infection. For patients who do not respond sufficiently to corticosteroids or who do not tolerate corticosteroids, adrenocorticotropic hormone or plasma exchange can be considered.
After the acute relapse is treated, consideration should be given to use of DMTs, which primarily target the inflammatory, demyelinating aspects of the disease. A list of DMTs for MS approved by the U.S. Food & Drug Administration (FDA) is presented in Table 2.
| Injectable platform therapies | Interferon beta-1a (Avonex, Rebif, Plegridy) Interferon beta-1b (Betaseron, Extavia) Glatiramer acetate (Copaxone, Glatopa) |
|---|
| Injectable therapy/monoclonal antibody therapy | Daclizumab (Zinbryta) ª |
| Oral therapies | Fingolimod (Gilenya) Teriflunomide (Aubagio) Dimethyl fumarate (Tecfidera) |
| Infusion therapies/monoclonal antibody therapy | Natalizumab (Tysabri) Alemtuzumab (Lemtrada) Ocrelizumab (Ocrevus) |
| Infusion therapy/chemotherapeutic | Mitoxantrone (Novantrone) |
ª Voluntarily withdrawn from the market in March 2018 due to concern about the benefit/risk profile, see https://www.fda.gov/Drugs/DrugSafety/ucm600999.htm
Current therapies target the immune dysfunction in MS and resultant neural tissue damage with the goal of preventing or at least reducing the long-term risk of clinically significant disability. Early treatment is key since it offers the greatest chance of preventing or delaying tissue injury and long-term disability. Although the underlying pathogenesis of MS still remains poorly understood, remarkable progress has been made in the development of drug therapies that inhibit disease activity. Using available options, including the advent of newer more effective drugs, there is the potential to achieve a disease-free status, characterized by the prevention of clinical relapses and disability progression and absence of new lesions on MRI. This widely accepted treat-to-target approach is known as “No Evidence of Disease Activity” (NEDA). Patients who achieve NEDA may have better long-term outcomes, although maintaining NEDA over the course of years or decades is challenging.
Escalation therapy and high-efficacy early therapy (HET) are 2 general management strategies to achieve NEDA in RRMS. In escalation therapy, the patient is initially started on a lower efficacy agent, such as one of the standard injectable DMTs. The rationale for using lower efficacy treatment is that such agents typically have a more desirable safety profile for better long-term safety overall. In the presence of disease activity, the patient is subsequently switched to higher efficacy treatment, often in a step-wise approach (eg, injectable → oral → infusion). HET is an alternative approach in which patients are started on high-efficacy therapy (eg, natalizumab and ocrelizumab) early in the course of their disease, and even as first-line agents. This approach may carry more risk in certain circumstances, especially given that patients will have more long-term exposure to drugs with significant immune-altering effects. However, the rationale behind this treatment strategy is that the benefits of early disease control outweigh the risks and may have longer-term benefit compared with escalation therapy. There are middle-ground approaches, too, that utilize oral therapies such as fingolimod and dimethyl fumarate as first-line treatments. Multicenter, prospective, pragmatic clinical trials are needed to address these important clinical questions.
It is important to note that all current MS therapies are preventative and not restorative. As the disease progresses, response to DMT typically declines. The key to successful treatment of MS is to slow the inflammatory process early in the disease. The therapeutic nihilism of the past should be replaced by aggressive treatment and monitoring, while carefully balancing the potential risks and benefits. Monitoring patients clinically and with surveillance MRI scans during treatment is important to detect non-responders and modify therapy accordingly.
It is likely that the accumulation of irreversible tissue damage limits the potential for benefit from DMT as the disease progresses. However, with better understanding of MS pathogenesis and identification of appropriate outcomes measures for progressive MS in clinical trials, the therapeutic landscape of DMT strategies for this disabling and neurodegenerative disease state is rapidly evolving and holds great promise for the future.
Four first-line injectable therapies, otherwise known as platform agents, are currently available in the US: intramuscular interferon (IFN) beta-1a (Avonex), subcutaneous IFN beta-1a (Rebif; Plegridy), subcutaneous IFN beta-1b (Betaseron, Extavia), and glatiramer acetate (Copaxone, Glatopa) (Table 3). The IFN medications are recombinant products with an amino acid sequence that is identical or nearly identical to that of human IFN beta-1. Glatiramer acetate is a random polypeptide based on the amino acid sequence of a myelin protein. All of these medications appear to modulate the immune response in MS, although glatiramer acetate and IFN beta medications probably work through different mechanisms.
| Drug (brand name) | Dose | Adverse Effects | Lab monitoring/risk mitigation |
|---|---|---|---|
| Interferon β-1a (Avonex) |
30 mcg intramuscular once weekly |
Injection site reactions Flu-like symptoms Lymphopenia Hepatotoxicity Exacerbation of preexisting thyroid disease |
Baseline complete blood count (CBC) and liver function tests (LFTs) Periodic CBC and LFTs every 3 to 6 months Baseline thyroid function assessment, then periodically with clinical symptoms |
| (Rebif) | 44 mcg subcutaneous 3 times weekly |
||
| (Plegridy) | 125 mcg subcutaneous every 2 weeks |
||
| Interferon β-1b (Betaseron)(Extavia) |
0.25 mg subcutaneous every other day |
||
| Glatiramer acetate Copaxone, Glatopa |
20 mg subcutaneous once daily |
Injection site reactions Post injection systemic reactions Lipoatrophy |
None |
Based on data from Oh J, et al.21
In randomized, placebo-controlled trials, all of these medications decreased the rate of clinical relapses by about 30%, decreased the severity of the relapses, and had beneficial effects on measures of disease activity on MRI.22-25 All of the platform medications are reasonably well tolerated, and 15 to 20 years of accumulated data and clinical experience suggest strong long-term safety. The platform therapies are similar in efficacy, and selection is generally based on physician and patient preferences and side-effect profile. Potential adverse effects of the IFN medications include hepatic and hematological toxicities, flu-like side effects, and worsening of headaches, depression, and spasticity. Glatiramer acetate may have the potential for more bothersome injection site reactions, particularly in thin patients. All 4 injectable platform therapies are appropriate first-line therapies in RRMS.
Daclizumab (Zinbryta, 150 mg once monthly subcutaneous injection) was voluntarily withdrawn from the market in March 2018 due to concern about the drug’s benefit/risk profile(https://www.fda.gov/Drugs/DrugSafety/ucm600999.htm). Approved by the FDA for relapsing forms of MS in May 2016, daclizumab is a humanized monoclonal antibody that binds to and blocks the high-affinity interleukin-2 receptor alpha chain, which inhibits T-cell activation and proliferation. Its clinical benefit is also thought to result from the expansion of a subset of regulatory natural killer cells.26
Noted adverse effects are hepatotoxicity, autoimmune hepatitis, cutaneous reactions, and autoimmune phenomena. Lab monitoring and risk mitigation strategies include
Randomized controlled trials (RCTs) of daclizumab found that it reduces the annualized relapse rate (ARR) by 54% compared with placebo27 and by 45% compared with active comparator intramuscular IFN beta-1a.28 The number of new or newly enlarging T2-hyperintense lesions was 54% lower with daclizumab than with intramuscular IFN beta-1a over a period of 96 weeks.28
Overall, the incidence of adverse effects was comparable between daclizumab and comparator treatments in phase 3 clinical trials.27,28 However, daclizumab is associated with a higher proportion of serious infections. Of these, urinary tract infections, upper respiratory tract infections, pharyngitis, and sinusitis are the most common adverse effects. Most resolve with standard treatments without subsequent complications. There was 1 patient treated with daclizumab who died from complications of a local psoas abscess. Other side effects include cutaneous reactions, 1 case of autoimmune hepatitis,29 and hepatotoxicity including 1 case of fatal fulminant hepatic failure.30
There are currently 3 oral DMTs approved by the FDA. These therapies include fingolimod (Gilenya), teriflunomide (Aubagio), and dimethyl fumarate (Tecfidera)
(Table 4).
| Drug (brand name) | Dose | Adverse effects | Lab monitoring/risk mitigation |
|---|---|---|---|
| Fingolimod (Gilenya) | 0.5 mg by mouth daily |
|
|
| Teriflunomide (Aubagio) | 7 mg or14 mg by mouth daily |
|
|
| Dimethyl fumarate (Tecfidera) | 240 mg by mouth twice daily |
|
|
CBC = complete blood count; ECG = electrocardiogram; LFTs = liver function tests.
Based on data from Oh J, et al.21
Fingolimod was approved in September 2010 as the first oral disease therapy for MS. Fingolimod acts by binding to the sphingosine-1-phosphate receptor on lymphocytes, which prevents egress of lymphocytes from lymph nodes. The sequestration of autoreactive lymphocytes prevents their recirculation to the CNS, thus inhibiting one of the primary steps in MS pathogenesis. Fingolimod crosses into the CNS and may have direct effects within the CNS, as well.
Clinical trials of fingolimod found that it reduces the ARR by 54% and disability progression by 30% in comparison to placebo, with MRI measures of disease activity supporting these clinical findings.31 In a head-to-head clinical trial against active comparator IM IFN beta-1a, fingolimod demonstrated a 33% reduction in ARR and a reduction in MRI disease activity, but no difference in disability progression.32
Most fingolimod-associated side effects are mild to moderate in severity and include upper respiratory tract infections, headache, diarrhea, and back pain. The most concerning adverse effects include cardiac events (bradycardia and atrioventricular block at treatment initiation), elevated liver enzymes, rare serious infections (eg, herpes virus infections), and macular edema. The development of these serious side effects during clinical trials led to strict FDA recommendations for close monitoring during first dose administration and risk factor mitigation strategies to reduce potential serious complications. These parameters include baseline complete blood count (CBC), liver function tests (LFTs), electrocardiogram, ophthalmological evaluation, and serum varicella virus immunoglobulin G titer prior to fingolimod initiation. First-dose administration is conducted under the supervision of a healthcare provider (either at home or in a medical center) where patients are monitored for 6 hours with hourly vital sign checks and a repeat electrocardiogram after 6 hours. Extended monitoring for a total of 24 hours is needed if bradycardia or QT prolongation is observed, and with other cardiac risk factors. Periodic labs including CBC and LFTs and ophthalmological reassessment are used for continued safety surveillance.
Cases of progressive multifocal leukoencephalopathy (PML) have been reported in association with fingolimod. PML is a serious viral infection of the brain, arising from the ubiquitous John Cunningham virus (JCV), which resides in the kidneys and bone marrow in about half of adults. The estimated rate of PML with fingolimod is about 1:10,000 overall, with a higher rate in those treated for more than 2 years.33
Teriflunomide was the second oral DMT approved by the FDA in September 2012. It is an active metabolite of leflunomide and acts by inhibiting the de novo synthesis of pyrimidine nucleotides through the inhibition of dihydroorotate dehydrogenase.34 It also inhibits T-lymphocyte activation and cytokine production in addition to cytostatic effects on proliferating B- and T-lymphocytes.35 Phase 3 trials of teriflunomide showed that it reduces the ARR by35 compared with placebo.36,37 However, a phase 3 trial comparing teriflunomide and subcutaneous IFN beta-1a showed them to have relatively similar efficacy.38
Teriflunomide is relatively safe and generally well-tolerated. There is no increased risk of opportunistic infections, and most of the adverse effects related to the medication are transitory. The most common adverse effects observed in RCTs and in clinical practice are mild to moderate in severity and include nasopharyngitis, gastrointestinal symptoms, decrease in hair density, mildly elevated LFTs, rash, and fatigue. Patients should be screened for tuberculosis before initiating therapy. Risk mitigation strategies include LFTs at baseline and every 6 months while on the medication and a baseline and 6-month CBC.39
Although teriflunomide was not carcinogenic in mice and rats, it was found to be mutagenic and resulted in embryo lethality in rats. Thus, it has a pregnancy category X designation. In this context, pregnancy must be excluded in all women of childbearing potential prior to treatment, and effective contraceptive methods must be employed for both women and men. An accelerated removal process is available for patients who become pregnant or desire to become pregnant (or father a child) while taking teriflunomide.
Dimethyl fumarate (DMF) is the third oral DMT that was approved in March 2013. It is a fumaric acid ester with mechanism of action in MS that is thought to be related to its effect on nuclear factor (erythroid-derived 2)-related factor 2.40 Fumaric acid esters have been used in Europe to treat psoriasis under the brand name Fumaderm and are known to have a relatively favorable safety profile.41 DMF demonstrated 48% to 53% ARR relative reduction compared with placebo,42,43 in addition to reduced MRI activity.40 There was a reduction in the risk of disability progression in one trial,42 although this finding was not significant in a second RCT.43
The most frequent adverse effects associated with DMF are gastrointestinal symptoms, including stomach pain, nausea, vomiting, and diarrhea. Gastrointestinal symptoms are generally more prominent during the first several weeks of treatment and usually improve significantly thereafter. Taking DMF with food and slower initial dose titration may offset potential gastrointestinal side effects. Transient skin flushing is also observed intermittently. Concomitant use of low-dose aspirin substantially reduces associated skin flushing.
Lymphopenia is a possible side effect without associated increased risk of serious infections. Cases of PML have been reported in association with dimethyl fumarate. The estimated rate of PML with dimethyl fumarate is less than 1:15,000 overall, with a higher rate in those with sustained lymphopenia (ie, < 500 lymphocyte count persisting for > 6 months). In this context, baseline and periodic CBC monitoring every 6 months is recommended surveillance measures while on DMF.
There are currently 3 infusion DMTs approved by the FDA to reduce disease activity in relapsing forms of MS and are considered highly effective therapies. These DMTs include natalizumab (Tysabri), alemtuzumab (Lemtrada,), and ocrelizumab (Ocrevus) (Table 5). Ocrelizumab is also the first DMT for treating patient with PPMS approved by the FDA.
| Drug (brand name) | Dose | Adverse effects | Lab monitoring/risk mitigation |
|---|---|---|---|
| Natalizumab (Tysabri) | 300 mg intravenous every 4 week |
|
Before infusion:
|
| Alemtuzumab (Lemtrada) | First course: 12 mg intravenous daily for 5 consecutive Second course: 12 mg intravenous daily for 3 consecutive days at month 12 from initial course |
|
Before Infusion:
|
| Ocrelizumab (Ocrevus) | 300 mg intravenous at week 0 and week 2, then 600 mg intravenous every 24 weeks |
|
Before infusion:
After infusion:
|
Ab = antibody; bid = twice daily; CBC = complete blood count; CMP = complete metabolic profile; CSP = cerebrospinal fluid; HIV = human immunodeficiency virus; JCV = John Cunningham Virus; LFTs = liver function tests; MRI = magnetic resonance imaging; PCR = polymerase chain reaction; PO = oral; SCr = serum creatinine; TB = tuberculosis; TSH = thyroid stimulating hormone; VZV = varicella zoster.
Natalizumab, approved by the FDA in November 2004, is a monoclonal antibody targeting the cellular adhesion molecule very late antigen-4. By blocking very late antigen-4, fewer inflammatory cells enter the brain and thereby blunt CNS inflammation typical of MS. Clinical trials of natalizumab showed that it reduces clinical relapses by 67% and new brain lesions by 83% in pivotal RCTs.45,46 Thus natalizumab is considered one of the most clinically effective DMTs for relapsing MS to date.
Natalizumab is relatively well-tolerated with mild headache, fatigue, anxiety, menstrual irregularities, peripheral edema, and routine infections (eg, upper respiratory infection and pharyngitis) occasionally observed. Infusion-related hypersensitivity reactions (eg, hives and pruritus) occur in 2% to 4% of patients and are thought to represent immune-mediated hypersensitivity reactions.47 Anaphylactic reactions are very rarely observed, but when observed, are typically during the second infusion. Patients who demonstrate a serious infusion reaction should discontinue natalizumab immediately and not be retreated.
The most concerning serious adverse effect of natalizumab is PML, which occurs at an overall incidence of 2.1 per 1,000 population.48-50 Three identified risk factors that substantially alter an individual’s risk of PML include duration of natalizumab treatment, prior history of immunosuppressive therapy, and serum anti-JCV antibody status. Patients with natalizumab treatment exceeding 60 months, prior use of immunosuppressant drugs, and positive serum anti-JCV antibody testing, carry the highest estimated risk for PML at 1:119 persons. Patients who are negative for JCV antibody have a low risk of PML, estimated at 1:14,285 persons.51
Because of PML, natalizumab was withdrawn from clinical use in February 2005 but received a second FDA approval in June 2006. Due to the serious potential for PML, natalizumab is generally reserved for patients with worrisome baseline disease activity or negative prognosticators or both, or patients who respond sub-optimally or do not tolerate other MS therapies. However, the growing experience of PML risk stratification suggests that in subjects with persistently negative JCV serology, natalizumab can be considered as first-line therapy. Like with all medications, discussions with potential natalizumab recipients should review the risks and potential benefits of this medication.
In the context of natalizumab’s risk of PML, careful risk stratification prior to treatment initiation is recommended, which enables more informed clinical decision making. It appears that natalizumab-related PML has a better prognosis than PML in other settings, although fatalities or persistent deficits are common. Accelerated removal of natalizumab from the blood (ie, through plasmapheresis or leukopheresis) likely accelerates immune reconstitution and is recommended in patients with natalizumab-related PML.
Alemtuzumab, approved by the FDA in November 2014 for relapsing MS, is a humanized antibody that targets CD52, a cell surface protein expressed on T-lymphocytes, B-lymphocytes, natural killer cells, monocytes, and dendritic cells.52 Alemtuzumab induces rapid depletion of circulating T- and B-lymphocytes followed by repopulation that leads to a distinctive lymphocyte profile, including an increased proportion of regulatory T-lymphocytes and memory B- and T-lymphocytes. In contrast to the slow recovery of T-lymphocytes, B-lymphocytes return to baseline levels by 3 months, which may explain the occasional development of secondary humoral autoimmune disorders.53
RCTs of alemtuzumab showed that it reduces the number of clinical relapses versus active comparator, subcutaneous IFN beta-1a, by about 49% to 55%, in both treatment-naïve54,55 and treatment-experienced patients.56 Two of these trials showed reduction in the risk of confirmed worsening of disability, and all 3 showed slowing in progression of cerebral atrophy.
The most common adverse effect is infusion-related reactions (IRRs), which occurs in over 90% of patients treated with alemtuzumab without pre-medications.54,55 The most common symptoms are headache, rash, and pyrexia; which are mostly mild-moderate in severity. IRRs are mitigated by a pretreatment protocol with intravenous methylprednisolone and symptom management with antihistamine and antipyretics.
The principal adverse effect of alemtuzumab is secondary autoimmune disorders, which is theorized to be related to the distinct lymphocyte repertoire that develops following alemtuzumab exposure. Thyroid disease is the most common, occurring in up to 34% in patients followed over 7 years.57 Other rare secondary autoimmune adverse effects include immune thrombocytopenic purpura and antiglomerular basement membrane disease that led to renal transplant in a patient outside of clinical trials.58 In the extension phases of clinical trials through year 6, the peak of thyroid disease occurred during year 3, nephropathy during years 1 to 2, and immune thrombocytopenic purpura throughout the study period.59,60
The subsequent period of rapid lymphocyte depletion immediately following alemtuzumab exposure is associated with a mild increase in infections. Most of these infections are mild to moderate nasopharyngitis, urinary tract infections, and upper respiratory tract infections, although serious listeria and herpes viral infections can occur. Due to the risk of listeria and herpes viral infections, it is recommended that patients be treated with twice daily trimethoprim/sulfamethoxazole for 2 months and daily oral acyclovir for 1 year following each treatment course of alemtuzumab.61
Ocrelizumab, approved by the FDA in March 2017, is a humanized anti-CD20 monoclonal antibody that binds to an epitope that overlaps with that of rituximab. It is hypothesized that ocrelizumab has comparable efficacy of B-cell depletion to rituximab. In contrast, ocrelizumab is purported to have fewer side effects than rituximab because it is a more humanized antibody and thus produces greater antibody-dependent cellular toxicity and less complement-dependent cytotoxicity.62 Pivotal phase 3 trials (OPERA I and OPERA II) demonstrated a significant ARR reduction for ocrelizumab compared with subcutaneous IFN beta-1a by 46% to 47%.63 In a pooled analysis, there was a 40% risk reduction in time to 12-week confirmed disability worsening and a 40% risk reduction in time to 24-week confirmed disability worsening between ocrelizumab and subcutaneous IFN beta-1a.
The most common adverse effects associated with ocrelizumab are IRRs. They typically are mild in degree and managed by symptomatic therapy or slowing the infusion rate. There are no increased rates of serious adverse effects or serious infections.
Malignancy occurred in 0.5% of patients treated with ocrelizumab compared with 0.2% of subcutaneous IFN beta-1a treated patients in clinical trials.63 Two cases of breast cancer emerged in patients receiving ocrelizumab in 1 of the pivotal RCTs versus none in patients treated with subcutaneous IFNβ-1a.63 An additional 2 cases of breast cancer occurred during the open-label extension study, during which all patients received ocrelizumab. Given these small numbers, it remains unclear whether ocrelizumab treatment increases the risk of cancer.
Ocrelizumab may increase the risk of PML, but to date there have been no cases of PML directly attributable to ocrelizumab.
Cyclophosphamide, methotrexate, azathioprine, and cyclosporine have all been studied in small- to medium-sized trials. The Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and MS Council for Clinical Practice Guidelines have made recommendations regarding these therapies.64 Methotrexate, azathioprine, and cyclosporine were each found to be possibly effective (type C recommendation) in altering the course of disease, but cyclosporine was found to have an unacceptable risk/benefit ratio. In their review, pulse cyclophosphamide treatment did not alter the course of MS (type B recommendation), although a more recent clinical trial showed reduced relapses and MRI lesions in patients treated with cyclophosphamide.65 Given that there are more than a dozen therapies approved by the FDA for relapsing MS and relatively weak evidence supporting the efficacy of immunosuppressant therapies, they are infrequently used in the treatment of MS.
Mitoxantrone (Novantrone, 12 mg/m2 every 3 months; maximum lifetime dose of 140 mg/m2) is a chemotherapy medication with demonstrated efficacy in very active relapsing and progressive MS.66 Administration is via intravenous infusion every 3 months, although a monthly induction course is sometimes used in patients with very active disease. Infusion side effects include nausea and alopecia. Adverse effects include cardiotoxicity, acute myeloid leukemia, bone marrow suppression, and gonadal dysfunction. Because of the potential for long-term toxicities including cardiac injury and lymphoproliferative disorders, mitoxantrone is now rarely used in treating MS. Cardiac injury can occur years after completing therapy, which warrants surveillance echocardiogram or multigated acquisition scans prior to each infusion and after the medication has been discontinued.
Treatment of progressive MS is more challenging than relapsing MS. Previously, certain DMTs (eg, subcutaneous IFN beta-1a and mitoxantrone) approved for RRMS were also found in some trials to slow progression of disability in SPMS, though the effect was modest and seen primarily in those subjects with superimposed active inflammation. It is likely worthwhile to use the DMTs in progressive MS if there is evidence of persistent active inflammation (eg, clinical relapses or active lesions on MRI) and side effects are tolerated, but patients should be informed that these therapies have limited efficacy in slowing the gradual progression of disability seen in progressive MS.
An encouraging advancement in the treatment of progressive MS was seen with ocrelizumab in PPMS, where ocrelizumab treatment was associated with a 24% relative risk reduction of progression of disability compared with placebo at 12 weeks. However, this trial enriched patients with active inflammation by limiting enrollment to those under 56 years of age and disease duration less than 10 to 15 years.67 The benefit of ocrelizumab was markedly diminished in those without GdE lesions at baseline and those aged over 50 years.68 The efficacy of ocrelizumab in PPMS patients over age 55 is unknown. Accordingly, the benefit of ocrelizumab on the underlying progressive aspects of PPMS appears to be limited.
Positive phase 2 and 3 clinical trial findings for high dose biotin69,70 and siponimod71,72 for progressive MS have been reported. These advances are encouraging for the future development of neuroprotective and neurorestorative agents.
Broadly speaking, it is appropriate to consider changing DMT for 3 reasons: intolerable adverse effects, safety concerns, and breakthrough disease. With respect to adverse effects, every attempt should be made to manage them symptomatically. Of paramount importance is the patient’s tolerance of and adherence to DMT. It is important to address poor adherence, as it can lead to higher relapse rates and disease progression. If the patient remains intolerant of adverse effects, switching to an alternative agent is advisable.
With the emergence of newer therapeutics with various mechanisms of action and risk profiles, it is imperative that safety is monitored over the course of treatment. If at some point during treatment, the perceived benefits of the DMT no longer outweigh potential risks, then switching to a different agent should be considered. For example, in a patient treated with natalizumab who seroconverts to a highly positive JCV antibody titer, an alternative strategy without similar safety concerns, such as ocrelizumab, should be considered.
Despite its frequency in routine clinical practice, there is no consensus on the optimal approach to either defining or managing breakthrough disease. Breakthrough disease is generally defined as continued clinical or radiographic evidence of inflammatory disease activity despite treatment with an established DMT. Continued clinical relapses or new MRI lesions, particularly after 6 months of treatment with an established DMT, typically constitutes breakthrough disease. The severity of relapses, their subsequent recovery, and the number and size of new or active MRI lesions all contribute to defining when patients are considered to have sufficient breakthrough disease activity to merit changing therapies. Continued surveillance of clinical and radiographic measures of disease activity is important throughout treatment. In general, patients are seen clinically every 3 to 12 months, with repeat brain MRI every 6 to 12 months, depending on the patient’s baseline disease status and DMT.
Novel methods for longitudinal assessment of neurological performance and quality of life metrics for patients with MS are available for clinical use. At Cleveland Clinic, the Multiple Sclerosis Performance Test is used prior to each visit with the healthcare provider.73 Completed by patients using an iPad, it includes neuro-performance testing that objectively measures walking speed, manual dexterity, low contrast visual acuity, and cognitive processing speed. Patients also complete questionnaires that screen for depression and evaluate patients’ impression of his or her overall health. These longitudinal data are used to carefully monitor patient function and may detect changes that would otherwise escape notice during traditional neurological assessments.
Besides neurologic disability, MS can produce a variety of other symptoms that can interfere with daily activities. Identification and treatment of these symptoms should be considered throughout the disease course (Table 6). Specific recommendations for management of fatigue and urinary dysfunction have been outlined by the Multiple Sclerosis Council for Clinical Practice Guidelines. Aggressive evaluation and treatment for these and other symptoms of MS can significantly improve quality of life and are an important component of long-term management of patients with MS.
| Symptom | Pharmacotherapy (brand name) | Miscellaneous |
|---|---|---|
| Fatigue |
|
|
| Depression |
|
Psychotherapy
|
| Bladder dysfunction |
Anticholinergic medications for treatment of overactive bladder symptoms: Oxybutynin (Ditropan)
|
Important factors to assess include:
|
| Bowel dysfunction |
Bulk-forming agents:
|
|
| Spasticity |
|
|
| Sexual dysfunction | Sildenafil(Revatio, Viagra) β |
|
α The sacral neurostimulation device precludes the ability to undergo MRI studies, limiting its use in patients with MS where monitoring MRIs are often an integral part of patient management.
β Evidence for efficacy for sexual dysfunction in women with MS has been negative.
In addition to conventional pharmacologic therapy, there is growing interest in the use of lifestyle strategies to support wellness and mitigate disease-related outcomes in MS. This interest is based on a growing appreciation of the role of certain comorbidities and lifestyle factors on disease activity, disability, mortality, and overall quality of life. For example, key observational studies suggest an association between vascular comorbidities (eg, hypertension, hyperlipidemia, and type 2 diabetes) and an increased risk of disability and mortality.74 While evidence from randomized clinical trials is limited, there is evidence to suggest benefit from vitamin D supplementation, tobacco smoking cessation, routine exercise, and maintenance of emotional well-being as adjunct therapies to DMTs.
MS is a heterogeneous disease with a variable clinical course. Patients can progress rapidly over several years to significant disability or may have a few relapses and then remain clinically stable for many decades. The accumulation of disability in MS is slower than previously thought and varies widely between individuals. Early studies reported a relatively quick progression from disease onset to walking with a cane, with a median time of about 15 years.75 However, more recent natural history studies reported a longer time to reaching this disability milestone, with a median time from onset to cane of about 30 years. Likewise, in PPMS, early studies reported short median time from disease onset to cane of less than 10 years, whereas more current studies showed that median time is closer to 15 years.75 The advent of effective immunomodulating therapy for relapsing MS may in part explain a better long-term prognosis, but newer diagnostic criteria may have increased inclusion of MS patients with mild disease as well.
It is difficult to predict which patients will progress and which patients will remain relatively stable over time. Although there are clearly patients in whom the disease remains relatively mild, it is very difficult to predict which patients will eventually follow this course. There are several prognostic factors for unfavorable clinical outcomes. Older age at onset, Black race, Hispanic ethnicity, and initial symptoms involving cerebellar, spinal cord or pyramidal systems, and higher initial clinical activity (eg, high attack frequency and increased disability progression in the first 5 years) are all unfavorable prognostic factors.76 Smoking and low serum vitamin D levels have also emerged as additional predictors of poor long-term outcome. Prognostic radiologic measures include brain and spinal cord atrophy and number of GdE lesions. MRI measures are also useful tools when evaluating the effect of MS therapies and should be used routinely for DMT monitoring.76
Pregnancy does not seem to have a detrimental effect on the overall disease course of MS. In general, DMTs are not recommended during pregnancy, so efficient family planning with the help of the obstetrician can help minimize the amount of time the patient is off DMT. Pregnancy during MS is associated with a decreased incidence of relapses, but there is a rebound in relapse frequency in the postpartum period.78 Relapses during pregnancy can be treated with short courses of high-dose corticosteroids if needed, though it is preferable to not treat mild relapses since adverse effects to glucocorticoids can be seen. A mid-pregnancy visit with the treating neurologist is recommended for postpartum planning. It is also generally recommended that patients who were previously treated with DMT prior to pregnancy resume treatment immediately postpartum unless they plan to breastfeed. If breastfeeding is pursued, cranial MRI 2 months after delivery for disease surveillance is appropriate. If there is evidence of active disease, the benefits of breastfeeding should be balanced with the need to resume DMT.
Unfortunately, no DMT is proven to be safe during pregnancy or while breastfeeding, and so they are generally not recommended.79 Most of the available DMTs, including interferon beta, daclizumab, fingolimod, natalizumab, and alemtuzumab are pregnancy category C. Glatiramer acetate is pregnancy category B and is the safest DMT to use in women who need to continue DMT. The potential impact of brief exposures to DMTs (ie, during the first few weeks of pregnancy, before pregnancy is recognized) is relatively unknown, but appears to be minimal. Accordingly, although women who become pregnant while taking DMTs are generally recommended to discontinue DMTs, they can be reassured that the potential impact on their pregnancy is very low. As stated above, teriflunomide is pregnancy category X and should not be used in women of childbearing potential without effective contraception and counseling.
The effect of vaccines on MS has been studied very carefully and there appears to be no adverse effect of vaccines on the course of disease.80 Vaccines can be given safely in patients with MS and should be administered when clinically indicated, unless patients are on specific medications with an impact on response to vaccination. Inactivated vaccines are generally preferred, including in patients taking DMTs. Live attenuated vaccines are generally not recommended for a person with MS because of their theoretical ability to stimulate MS inflammation, although there is no compelling evidence showing an increased risk in the MS population of live attenuated vaccines at this time.