Published: June 2016
Crystal-induced arthropathies are a group of disorders that involve deposition of crystals in joints and soft tissues, resulting in articular and periarticular inflammation and injury. Two types of crystals — monosodium urate (MSU) and calcium pyrophosphate dihydrate (CPPD) — are commonly involved in gout and CPPD disease, which are described in this chapter.
Gout is a crystal-deposition disease caused by the super saturation and precipitation of MSU crystals in tissues resulting in inflammation and tissue damage. Gout is characterized by acute and subacute attacks. The clinical course of gout can be summarized into three stages:
Hyperuricemia is the underlying metabolic aberrancy in gout and is defined as a serum urate level >6.8 mg/dL, a level at which urate precipitates into MSU crystals.
Gout is a relatively common disease, with a higher prevalence in men than in women. Between 3 and 8 million American adults (about 3% of the population) are estimated to have gout.1-3 The estimated prevalence is nearly 7% among men older than 65 years.4
Overall, the prevalence of gout has been increasing.5 Factors contributing to this rise include increases in alcohol use, purine-rich diets, rates of obesity and the metabolic syndrome, and use of diuretic agents.6
There is a link between elevated levels of serum urate and the incidence of gout. Patients with serum urate levels ≤7 mg/dL have an annual incidence below 1%; for patients with serum urate levels ≥9 mg/dL, the incidence is more than 5%.7
Gout is an inflammatory crystal arthropathy resulting from the pathogenic effect of MSU crystals in the joints and soft tissue. Uric acid in body fluid at pH 7.4 exists in the urate form. Thus, when referring to uric acid in physiologic fluid, it is preferable to use the term urate.
Uric acid comes from the metabolism of purine nucleotides. Purine metabolism leads to inosine then hypoxanthine. Hypoxanthine is metabolized to xanthine, which is metabolized to uric acid. These two last steps are catalyzed by the enzyme xanthine oxidase, which is the major site for pharmacologic intervention by allopurinol. In humans, uric acid is the final product; humans lack the ability to degrade urate further.
Minimal amounts of urate are eliminated through the urinary and intestinal tracts. If the body is unable to eliminate large burdens of urate, hyperuricemia develops. As urate levels increase and saturate the synovial fluid or soft tissues, crystals precipitate, leading to tissue damage and the development of tophi. The accumulation of urate crystals in soft tissues and joints activates monocytes and macrophages to clear the crystals by phagocytosis. This leads to the release of proinflammatory cytokines and chemokines into the surrounding area, triggering a cascade of acute inflammatory reaction and influx of neutrophils into the joints, resulting in swelling of the joint or soft tissue.
The mechanisms leading to the self-limited inflammatory process are not fully known. The innate anti-inflammatory processes, possibly mediated by anti-inflammatory cytokines, may interrupt the inflammatory process. The natural course of gout resolves spontaneously, on average, in 1 to 2 weeks.8,9
An additional proposed mechanism involves the role of an inflammasome and interleukin 1 (IL-1) in the pathogenesis of inflammation induced by MSU (the crystal in gout) and CPPD (the crystal in CPPD disease). Cryopyrin inflammasome detects MSU and CPPD crystals and activates IL-1, resulting in an inflammatory cascade. These IL-1–mediated inflammatory effects of MSU crystals could be blocked by IL-1 inhibitors. This pathway presents an opportunity to treat patients with gouty arthritis who are otherwise intolerant of or inadequate responders to standard anti-inflammatory therapies. Large randomized, controlled trials are needed to assess the efficacy and side effects of blocking IL-1 in this patient population.
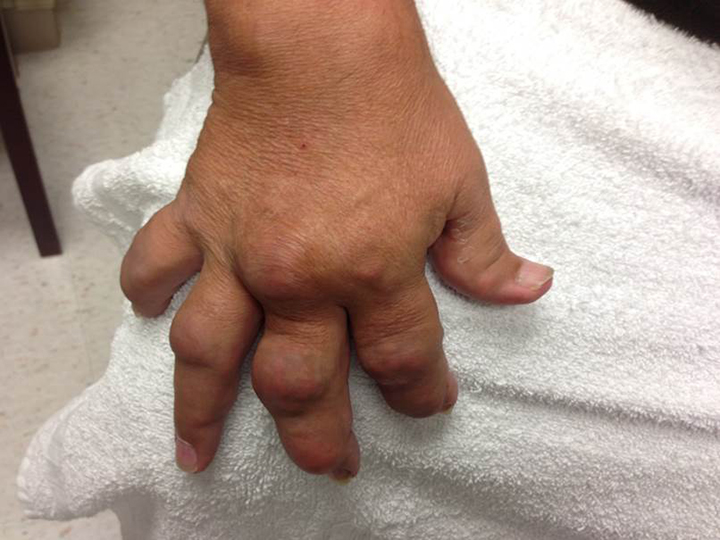
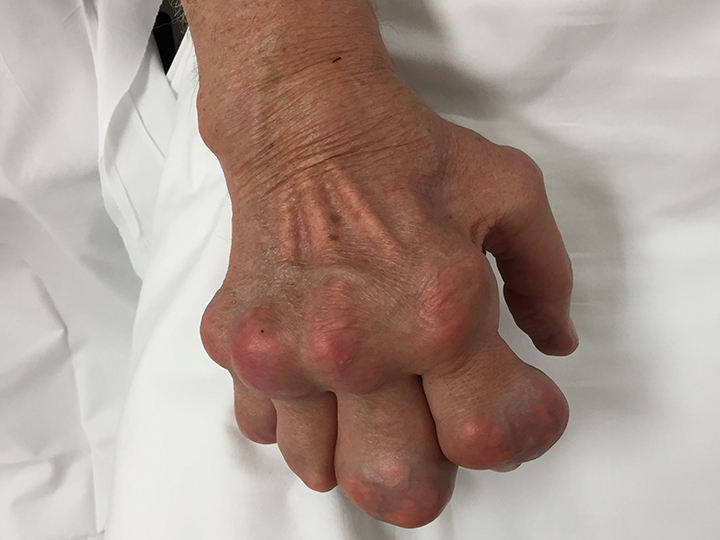
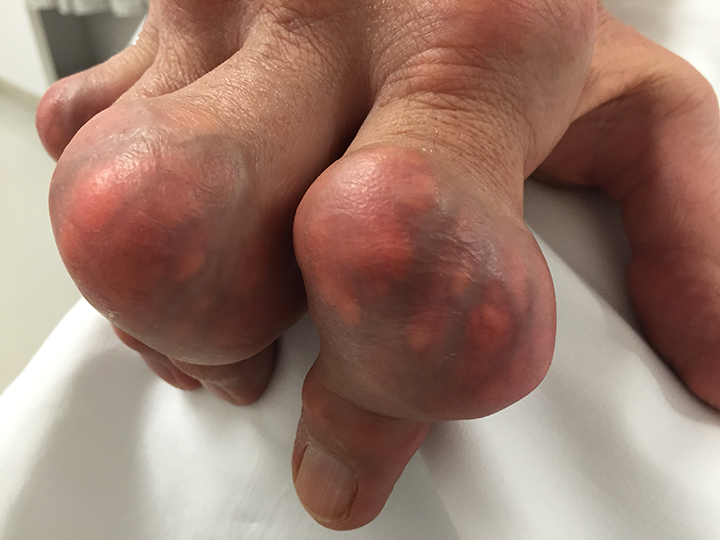
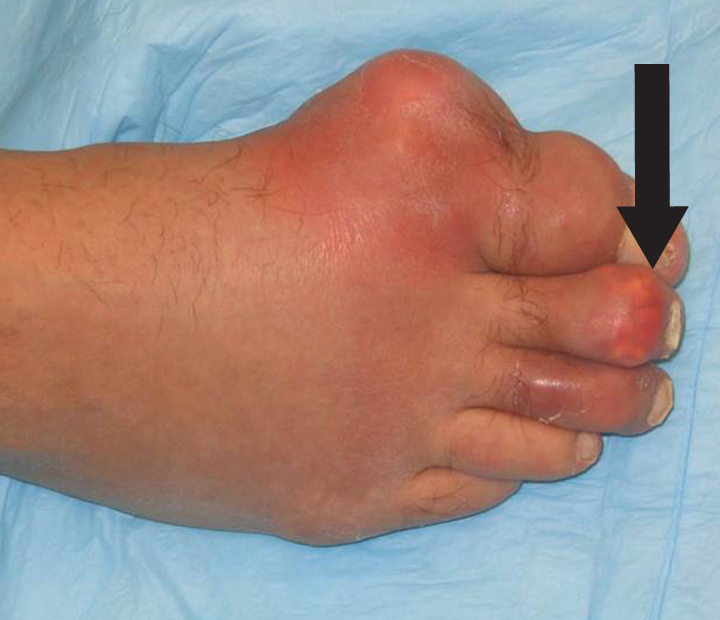
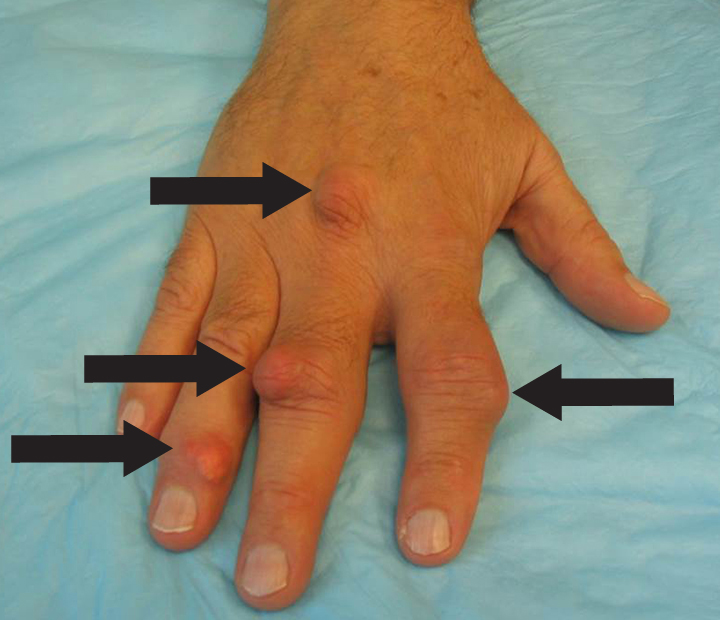
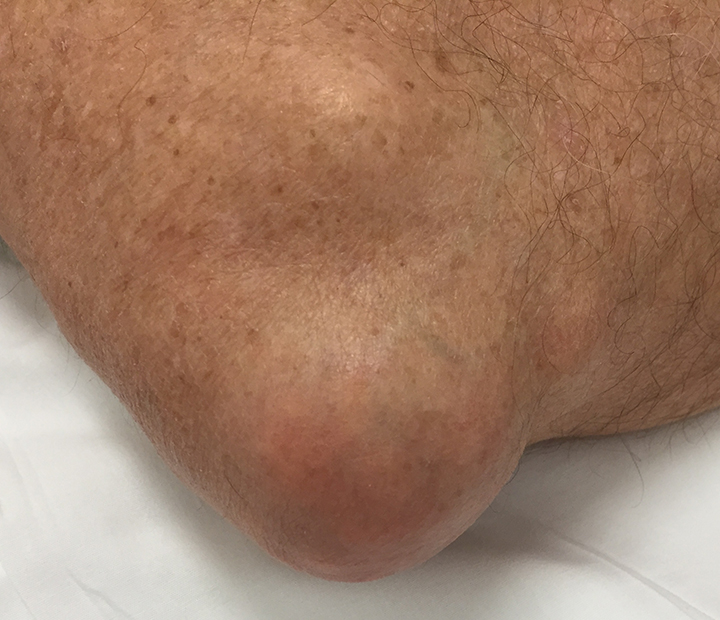
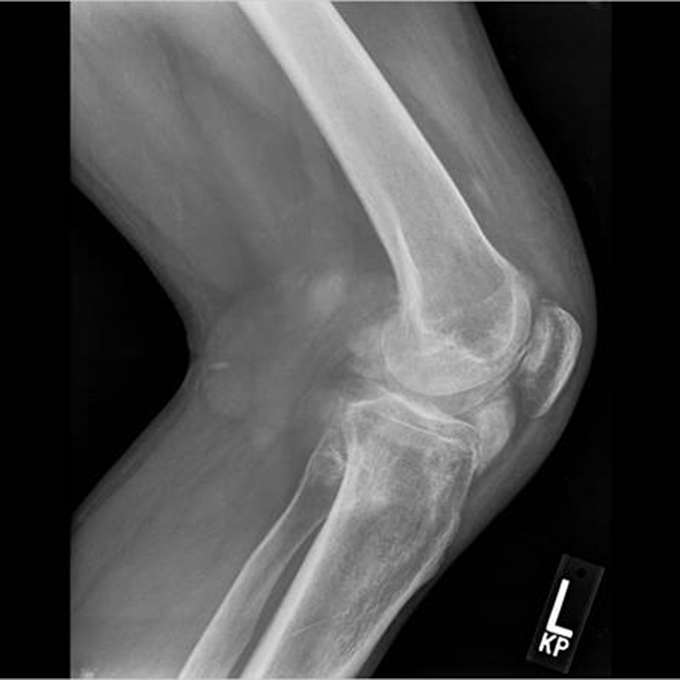
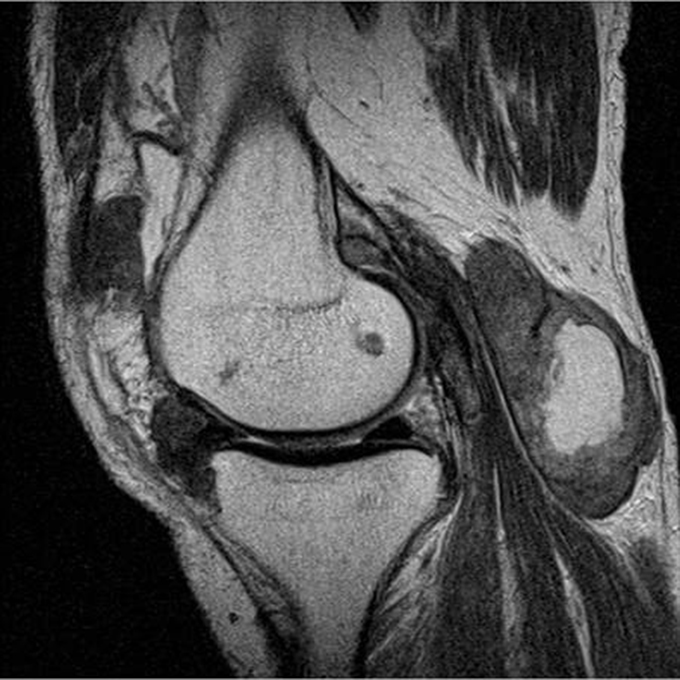
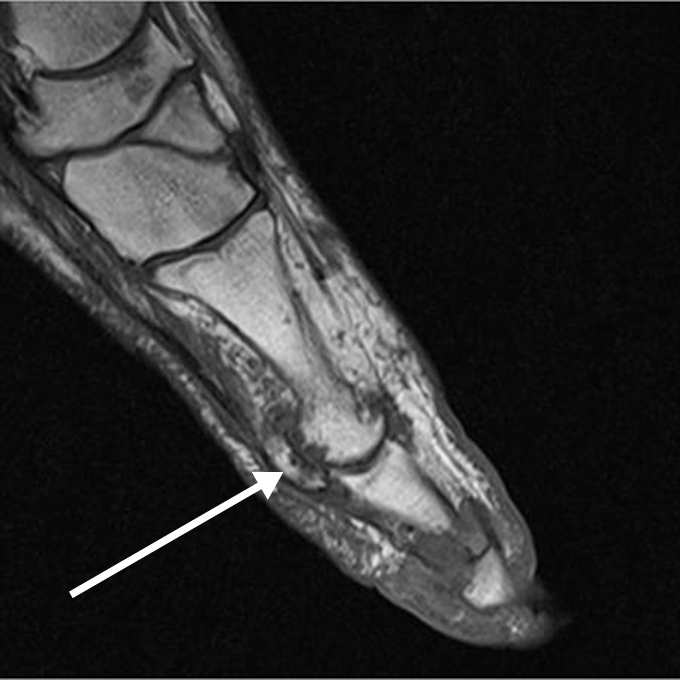
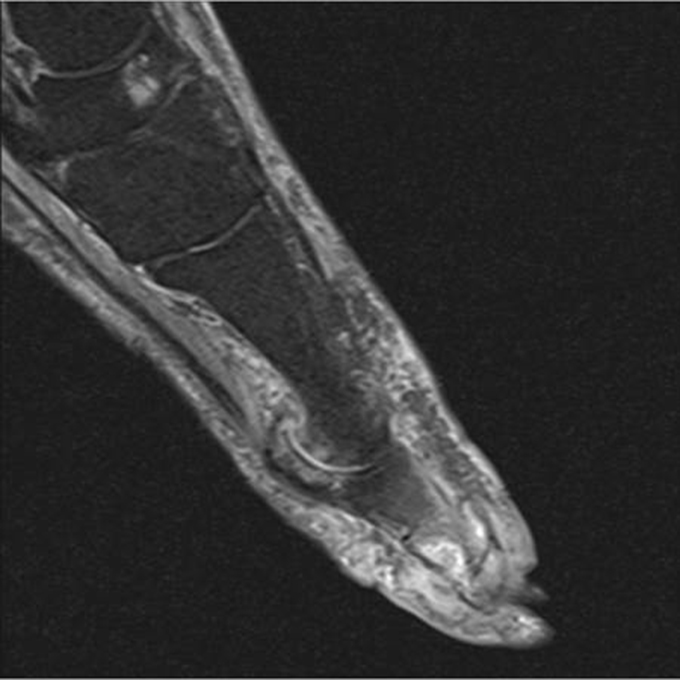
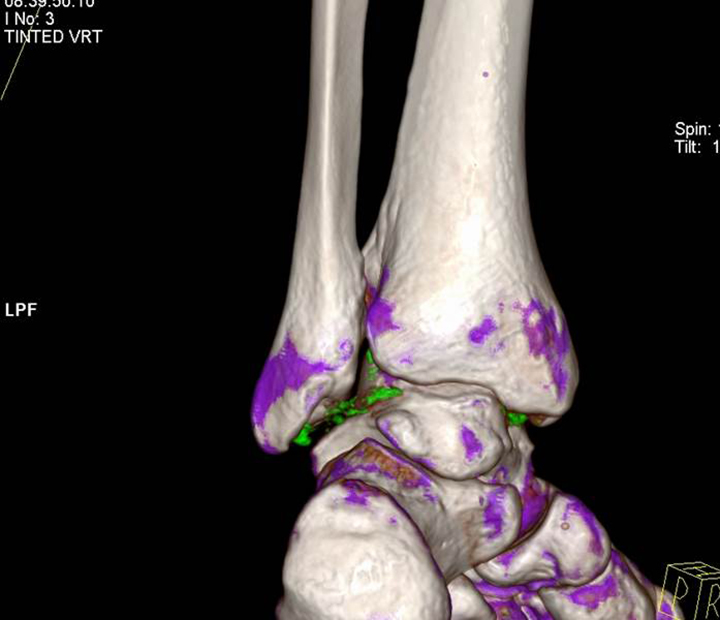
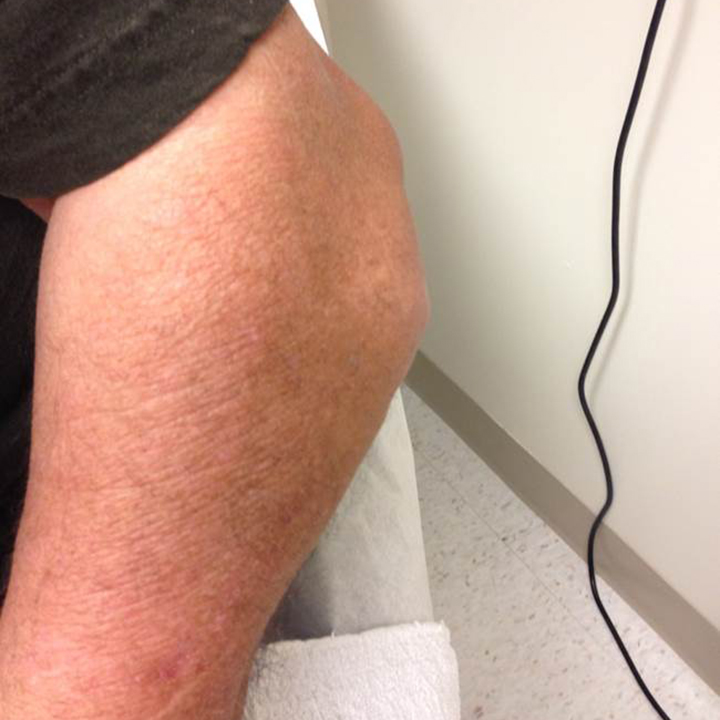
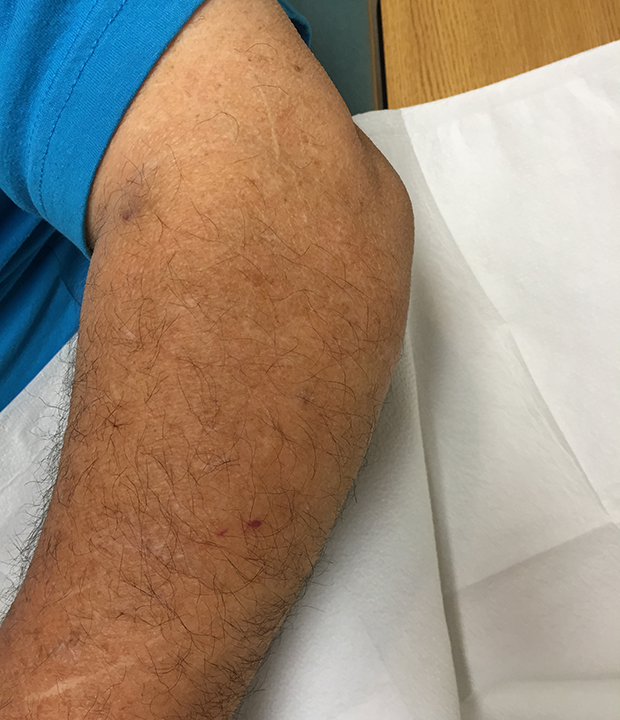
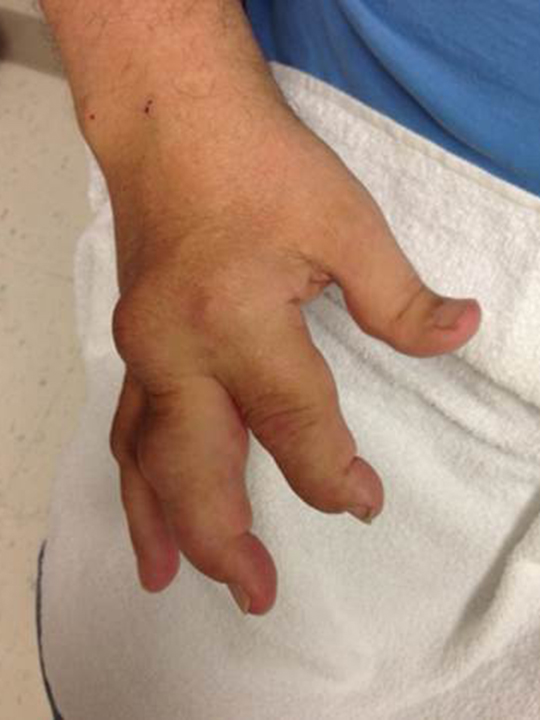
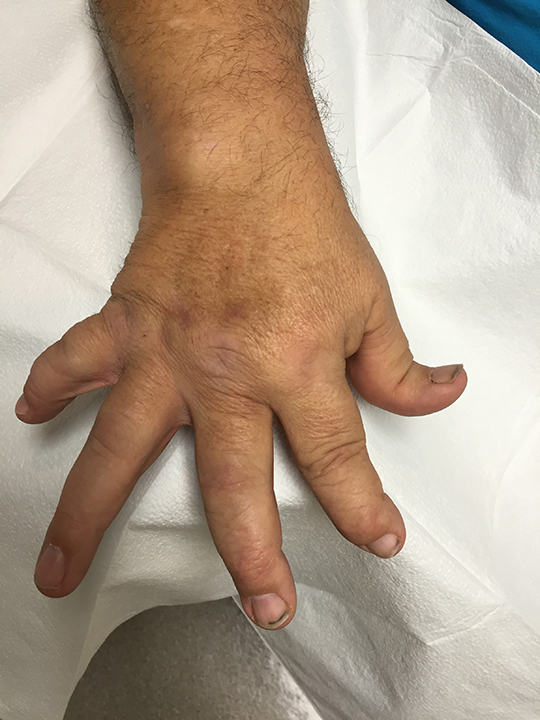
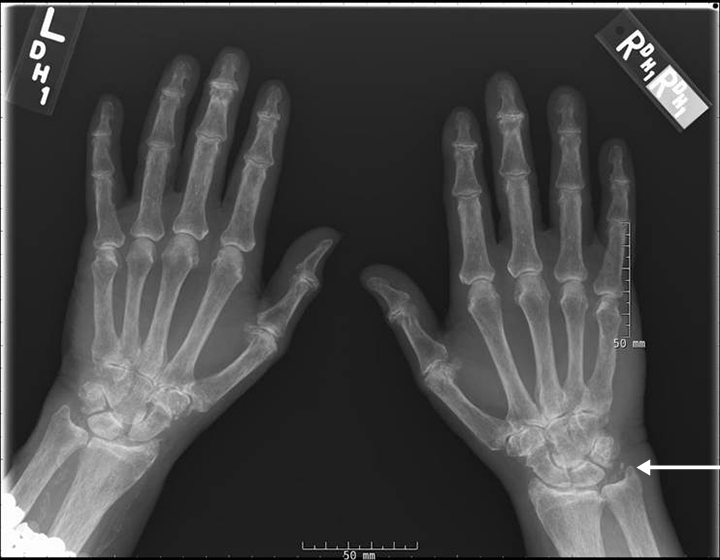
The acute inflammation of the joint or soft tissue associated with gout is clinically manifest as arthritis, direct soft tissue damage, and accumulation of MSU crystals (tophi) in soft tissue and bones (Figures 1-4). Hyperuricemia can cause uric acid nephrolithiasis and, possibly, nephropathy, if uric acid accumulates in the renal interstitium and tubules.
The arthritis in acute gout usually manifests as asymmetric monoarticular or oligoarticular inflammation, lasts 3 to 10 days, and resolves spontaneously. Eventually the attacks occur more frequently, last longer, and do not resolve completely, leading to chronic gouty arthropathy. Gouty arthropathy can lead to erosions and joint destruction. Gouty arthropathy is distinguished from rheumatoid arthritis by the absence of joint space narrowing and periarticular osteopenia.
In general, gout includes joint swelling in both the lower and upper extremities. Inflammation in the first metatarsophalangeal (MTP) joints is termed podagra, and it is highly suggestive of gout; however, any joint in the feet, ankles, knees, hands, wrists, or elbows may be involved. Acute gout can occur in the bursae, such as the olecranon or prepatellar bursae, where it causes bursitis, and can also occur in tendons such as the Achilles tendon and other soft tissue. Occasionally, a gout attack triggers a systemic inflammatory response manifesting with fevers, leukocytosis, elevated sedimentation rates, and elevated C-reactive protein (CRP).
Acute attacks can be precipitated by several factors. These include increased alcohol consumption (especially beer), diet (organ meat, shellfish), dehydration, trauma, and use of diuretics, cyclosporine, or urate-lowering drugs that can lead to sudden fluctuations in urate levels. Table 1 lists the common risk factors associated with gout.
| Hyperuricemia | Risk factors |
|---|---|
| Increased uric acid production |
|
| Reduced uric acid excretion |
|
When urate accumulates in a supersaturated medium, it can deposit in soft tissue or bones and form a tophus. Tophi can be present over the helices of the ears, extensor areas of the limbs, pressure areas such as the finger pads, and over the Achilles tendons. Most can be detected on a physical examination. On x-rays, they look like cystic or mass-like lesions. In general, tophi are radiolucent on x-rays, but when one occurs over a calcified nodule, it may be radiopaque.
If clinical suspicion of gout is raised, investigational studies are needed to confirm the diagnosis; elevated serum urate levels alone are not sufficient to make the diagnosis. The clinical presentation, medical history, and physical examination coupled with supportive evidence from additional testing, preferably synovial fluid analysis, can usually confirm the diagnosis. If inconclusive, additional studies may be needed, such as an x-ray, other imaging studies, or histopathology from surgical resections.
This analysis is considered the technical standard for evaluating patients with gout or acute CPPD arthritis. A synovial aspiration (ie, athrocentesis) for microscopic analysis should be obtained whenever feasible and examined grossly for color and turbidity. In general, transparent synovial fluid in the syringe is more suggestive of a noninflammatory condition, whereas fluid that appears turbid or purulent is more suggestive of inflammation or infection (eg, rheumatoid arthritis, gout, septic arthritis). Gross appearance alone, however, is not diagnostic.
To confirm or rule out infection, the fluid needs to be processed by Gram stain and culture. It is possible to have concomitant gout and septic arthritis. Microscopic examination can estimate the white blood cells (WBCs) count, which may be a useful adjunct in estimating the degree of inflammation present. With a gout attack, synovial fluid analyses may reveal leukocytosis, a nonspecific finding of inflammatory arthritis that includes infectious and crystalline causes.
Crystal analysis is done with compensated polarized light to detect and identify MSU crystals or CPPD crystals. Under compensated polarized light, MSU crystals are birefringent with strong negative elongation whereas CPPD crystals are weakly birefringent and rhomboid or rod shaped. Calcium pyrophosphate dihydrate crystals may not be as evident and can be missed, especially if the analysis is not done by a trained examiner. In addition to shape and birefringence, MSU and CPPD crystals differ in color depending on the axis of orientation with respect to the polarizer. When the axis of the MSU crystal is parallel to the polarizer, it appears yellow. When it is perpendicular, it appears blue. The CPPD crystal is the reverse. When it is parallel to the polarizer, it appears blue. When it is perpendicular, it appears yellow (Table 2).10
| Characteristic | Monosodium urate crystals | Calcium pyrophosphate dehydrate crystals |
|---|---|---|
| Birefringence | Strong | Weak |
| Shape | Needle-like, sharp edges | Rhomboid, rod-like |
| Color parallel to polarizer | Yellow | Blue |
| Color perpendicular to polarizer | Blue | Yellow |
Serum urate levels are not helpful indicators during acute gouty attacks because they can fluctuate from low to high. In chronic gout, a serum urate level is helpful to monitor and adjust the dose of urate-lowering therapy.
A complete blood count, serum creatinine, and liver function test are useful for evaluating other comorbid diseases, monitoring drug toxicity after initiating therapy, and assessing contraindications to a specific drug. For example, if tests show evidence of bone marrow suppression, advanced renal failure, or rhabdomyolysis, colchicine is not indicated. If tests show renal insufficiency, therapy with uricosuric medications (eg, probenecid) would not be effective because these medications require functional renal system to excrete excess urate.
A 24-hour urine test for uric acid levels is necessary when considering a uricosuric agent. Because results are partly affected by diet, it is best to repeat the test on two separate occasions. If the 24-hour urine levels are abnormally elevated, then uricosuric agents should not be used because of the increased risk of urate stones.
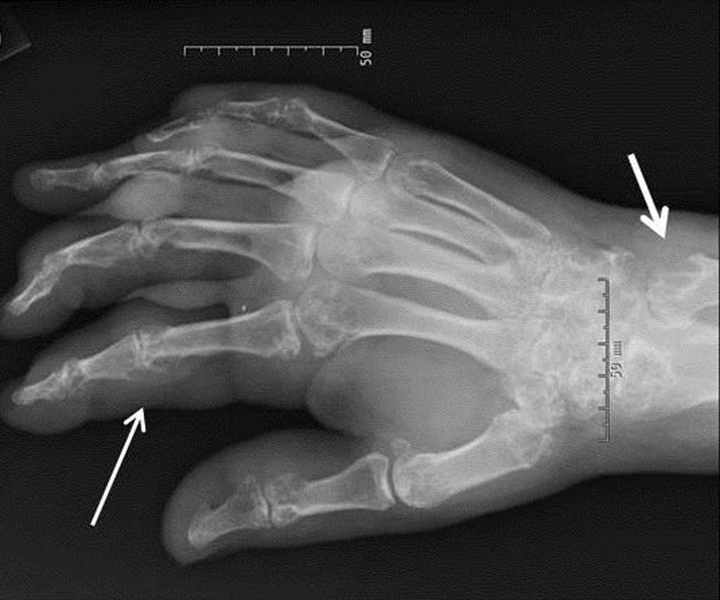
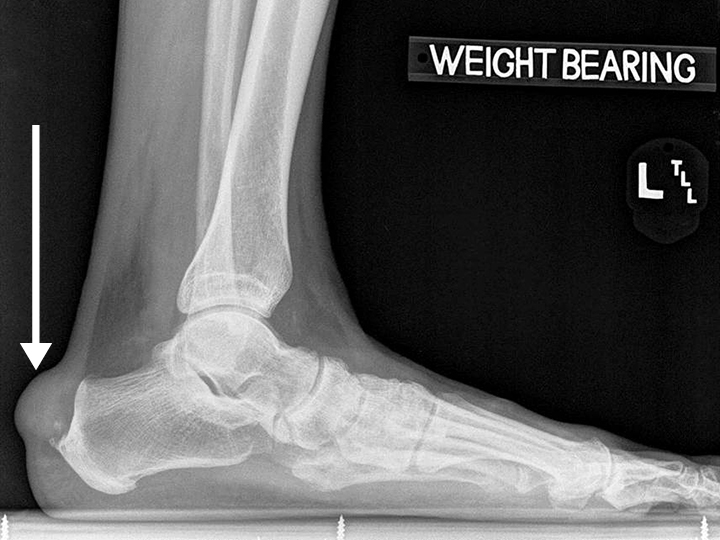
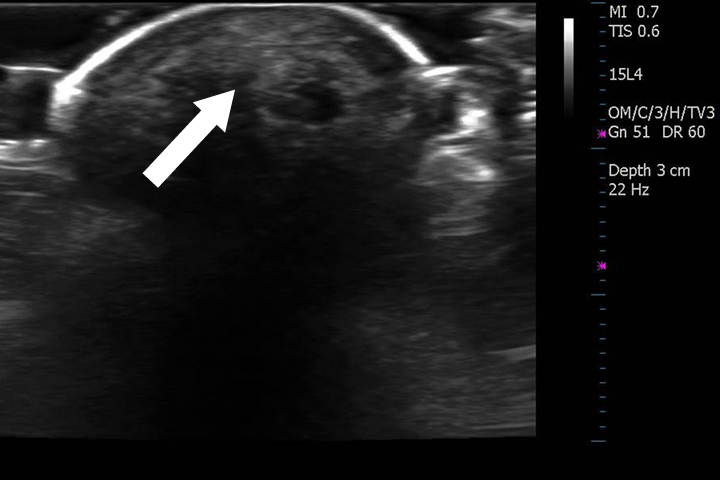
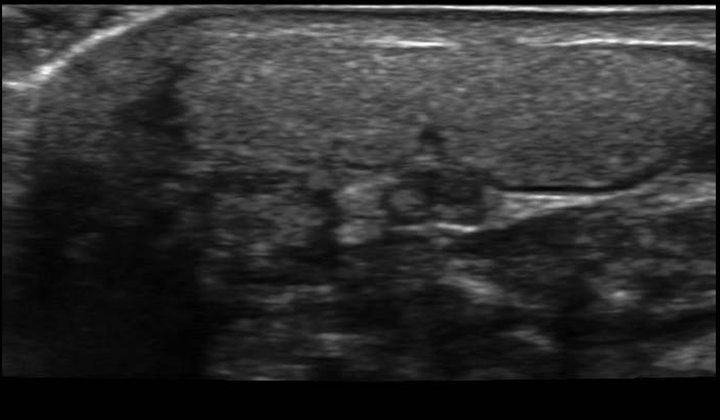
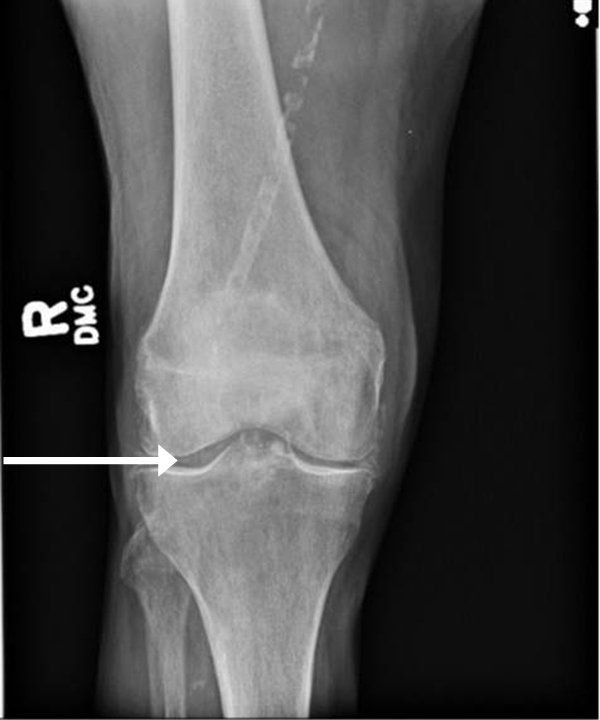
Arthropathy changes take years to develop, but evidence of erosions can appear much quicker. Crystal deposition can be detected by imaging, such as high-resolution ultrasound, before they can be seen on plain x-rays. Areas most commonly affected are the feet, ankles, hands, wrists, and elbows. On imaging studies, well-defined erosions with sclerotic margins and overhanging edges are typical of gout (Figures 5-6). Lack or absence of joint space narrowing and periarticular osteopenia can differentiate gout from rheumatoid arthritis.11
Other imaging modalities that have value in the investigation of gout and CPPD disease include high-resolution ultrasound, dual-energy computed tomography (CT) scan, and magnetic resonance imaging (Figures 7-11). In some studies, high-resolution ultrasound imaging and dual-energy CT were superior to plain radiographic imaging and, in certain cases, to synovial fluid analysis.12-14
The differential diagnosis is needed to evaluate for diseases and conditions with signs and symptoms that can mimic gouty arthritis. Various conditions can result in monoarticular arthritis, including infection, acute CPPD arthritis, other crystalline deposition disorders, atypical rheumatoid arthritis and palindromic rheumatism, other monoarticular arthritis, sarcoid arthritis, trauma, and fracture. Most have features that can be distinguished from gout by the clinical history, physical examination, and synovial fluid analysis. Thus, the differential diagnosis begins with a thorough medical history and clinical presentation. If no history of trauma or fracture is found, then a synovial aspiration of the joint is appropriate. In some cases, radiographic studies may be needed.
Once the diagnosis is confirmed, it is essential to exclude secondary causes, evaluate for known comorbidities associated with gout, and check for use of medications that increase the risk for hyperuricemia (eg, diuretics, cyclosporine, tacrolimus). Additional factors that increase the risk include excessive alcohol intake, diet rich in meat, and the existence of the metabolic syndrome, obesity, hypertension, hyperlipidemia, or renal disease (see Table 1). Comorbidities should be managed as clinically indicated.
Patient education is essential for patient adherence to therapy and success towards the prevention and management of gout. Patients should be informed about gout and its associated triggers and risk factors, as well as advice on lifestyle modifications (eg, avoid alcohol intake, limit consumption of meats, especially organ meats and shellfish, and ensure adequate hydration) and ways to alleviate the urate load and subsequent health risk.
Treatment depends on the clinical presentation and findings. The goal is to treat acute attacks, prevent future attacks, and prevent chronic joint and soft tissue damage caused by chronic tophaceous gouty arthropathy. Long-term gout therapy focuses on reducing serum urate levels by using xanthine oxidase inhibitors (ie, allopurinol, febuxostat) or the uricosuric agent probenecid. Because these agents are known to precipitate acute gout attacks, low-doses of colchicine are recommended for prophylaxis, usually lasting 6 months. The American College of Rheumatology (ACR) has created guidelines to help health care providers manage gout and hyperuricemia.15,16
Hyperuricemia and gout can be divided into three stages:
Treatments for each stage are discussed in the following sections. Table 3 shows the drugs commonly used to treat gout.
| Urate-lowering drugs | Generic (Brand) names | Notes |
|---|---|---|
| Xanthine-oxidase inhibitors | Allopurinol (generics available) Febuxostat (Uloric) |
|
| Uricosuric drugs | Probenecid (generics available) Lesinurad (Zurampic) |
Lesinurad must be used with a xanthine-oxidase inhibitor. |
| Uricase | Pegloticase (Krystexxa) Rasburicase (Elitek) |
Rasburicase is not approved by the FDA for gout. |
| Acute gout attacks | ||
| Nonsteroidal anti-inflammatory drugs | Diclofenac Ibuprofen Indomethacina Naproxena |
Commonly used nonsteroidal anti-inflammatory drugs; not a comprehensive list. |
| Colchicine | Colchicine (Colcrys, Mitigare) | No generic colchicine approved for gout but some used off-label. |
| Glucocorticoids | Prednisone Methylprednisolone |
|
| Interleukin inhibitors | Anakinra (Kineret) Canakinumab (Ilaris) |
|
aProphylaxis.
Data are insufficient to support treatment of asymptomatic hyperuricemia with hypouricemic agents. In general, initiating therapy in asymptomatic persons is not recommended, but investigating underlying comorbid conditions and addressing lifestyle factors may be appropriate.
Acute gout attacks can be managed with nonsteroidal anti-inflammatory drugs (NSAIDs), colchicine, or corticosteroids (intra-articular injection or systemic). All three agents are appropriate first-line therapy for acute gout. Therapy should be initiated within 24 hours of onset. The drug selection is dictated by the patient's tolerance of those medications and the presence of any comorbid diseases that contraindicates the use of a specific drug. For patients with severe or refractory gout attacks, practitioners can try combining agents. If all of these medications are contraindicated in a patient, narcotics may be used short term to relieve pain until the acute attack has resolved. Long-term use of narcotics should be avoided.
Nonsteroidal Anti-inflammatory Drugs. Although indomethacin has been traditionally used for acute gout, most other NSAIDs can be used as well. These drugs provide rapid symptomatic relief within the first 24 hours. As an example, indomethacin can be given at a daily dose of 150 mg (in three divided doses) for the first 3 days then 100 mg (in two divided doses) for 4 to 7 days.
Use of NSAIDs should be avoided in patients who are intolerant of such medications or who have other comorbid conditions contraindicating their use. Avoid NSAIDs in patients at risk for gastrointestinal (GI) bleeding, GI intolerance, or gastropathy; renal failure; hepatic failure; congestive heart failure; asthma; or hypersensitivity to NSAIDs. Avoid NSAIDs during the third trimester of pregnancy.
Colchicine. Colchicine works as an anti-inflammatory agent by blocking microtubule assembly in neutrophils, which attenuates phagocytosis and the transport of MSU crystals to the lysosome. Colchicine also impedes the activation of neutrophils in the vicinity of MSU crystals by blocking the release of chemotactic factors, thus diminishing recruitment of polymorphonuclear leukocytes to the inflamed joint.
Colchicine offers the best response when initiated within 48 hours of acute gout onset. Patients usually notice improvement within 24 to 48 hours of initiating therapy. During acute gout, oral colchicine can be started at 0.6 mg three or four times daily (preferably taken every 8 hours or in doses separated by at least 1 hour) for 2 days, then decreased to twice daily. Once gout symptoms resolve, colchicine can be stopped; however, it can be continued at a dose of 0.6 mg every 12 hours to prevent further attacks.
The adverse effect of diarrhea may limit dosing in some patients. Colchicine should not be prescribed if diarrhea develops. Colchicine requires dose adjustment in patients with decreased renal function and should not be used in patients on dialysis.
Other adverse effects of colchicine include abdominal cramps, bone marrow suppression, axon-loss neuropathy, myopathy (especially in renal insufficiency), potential liver toxicity, arrhythmia, shock, and skin rash (uncommon). Use caution in patients with biliary obstruction, hepatic failure, or renal insufficiency or end-stage renal disease and in pregnant women, patients with neutropenia, transplant patients taking cyclosporine, and patients experiencing renal function impairment or on statin therapy (risk of rhabdomyolysis). Concomitant use of colchicine with cyclosporine can lead to rapid-onset myopathy and increased myelosuppression.
Corticosteroids. In the case of systemic infections or septic arthritis, steroids should be avoided if possible. Corticosteroids may be used locally as an injection or systemically (orally, intramuscularly, or intravenously). Corticosteroids are usually very effective, and response is noticed within 24 hours of beginning therapy.
Oral corticosteroids can be as a methylprednisolone pack or prednisone starting at 40 mg (for example) or less, with a gradually tapering dose. Systemic steroids are the preferred agents in patients with renal failure in whom NSAIDs and colchicine are contraindicated. Local steroid injections may be the best alternative route of administration in patients who are unable to tolerate systemic therapy. Steroids, however, are not usually first-line therapy because of their potential adverse effects such as hyperglycemia, GI disease, weight gain and fluid retention, muscle weakness, immune suppression, and long-term effects such as those on bone resorption. These potential adverse effects need to be considered in the treatment decision.
The main treatment goal is to decrease urate levels to less than the level of precipitation, which is 6.8 mg/dL. The therapeutic target level should be urate levels <6 mg/dL to decrease the risk of a gout attack and formation of tophi, though urate levels of 5 mg/dL are preferred to decrease the formation of tophi.
Two pharmacologic routes are used to achieve low urate levels:
The decision to initiate hypouricemic therapy needs to be individualized, taking into consideration factors such as the absence of definite reversible causes of hyperuricemia, the number of attacks (two or more acute gouty attacks), the degree of hyperuricemia, and the presence of tophi. Treatment with a hypouricemic agent is usually lifelong and, thus, patient adherence is crucial.
During treatment with a urate-lowering agent, urate levels fluctuate, leading to increased risk of gout attacks. As a result, hypouricemic therapy is usually started only after the acute attack has completely resolved.
Also, prophylactic agents (colchicine, NSAIDs, or systemic steroids) should be initiated concurrently with, or prior to starting, urate-lowering therapy to decrease the risk of recurrent flares. Colchicine is the most popular prophylaxis therapy. Research suggests beneficial outcomes when treating patients with colchicine during initiation of urate-lowering therapy. Prophylactic therapy is best if continued for 6 months to 1 year, when possible.17,18 In patients who are unable to take colchicine, therapy with NSAIDs or low-dose steroids may be considered.
Allopurinol. Allopurinol inhibits xanthine oxidase, the enzyme responsible for the conversion of hypoxanthine to xanthine to uric acid. Steady doses of allopurinol have been shown to decrease serum urate levels.19 Before starting allopurinol, a thorough discussion with the patient is necessary regarding potential adverse effects. The patient must be cautioned about early signs and symptoms of hypersensitivity reactions.
Potential adverse effects of allopurinol include rash, nausea, vomiting, renal failure or impairment, and, less commonly, nephrolithiasis, bone marrow suppression, angioedema, bronchospasm, exfoliative dermatitis, pancreatitis, hepatitis, peripheral neuropathy, Stevens-Johnson syndrome, and toxic epidermal necrolysis.20 Allopurinol hypersensitivity syndrome can manifest with erythematous rash, fever, hepatitis, eosinophilia, or acute renal failure. Allopurinol hypersensitivity is a serious and potentially life-threatening reaction to allopurinol. If hypersensitivity is suspected, the drug should be discontinued immediately and the patient should be followed closely for failure of symptoms to resolve and for any progression of symptoms. Hypersensitivity can occur in patients with renal insufficiency; therefore, a low starting dose, 25 to 50 mg/day, is recommended in this patient population.
Drug interactions occur with oral anticoagulation (such as warfarin), azathioprine, mercaptopurine, cyclophosphamide, cyclosporine, and iron supplements. Allopurinol, azathioprine, and mercaptopurine share the same enzyme, xanthine oxidase, and could therefore increase the levels of those drugs, leading to exaggerated marrow suppression. Caution is necessary with the use of allopurinol in patients treated with cyclosporine, because it can increase the serum levels of cyclosporine.
American College of Rheumatology guidelines recommend screening for rapid polymerase chain reaction-based HLA-B*5801 before starting allopurinol in populations at high risk for severe allopurinol hypersensitivity reaction.15
The recommended starting dose is 100 mg/day, increasing gradually every 2 to 3 weeks to reach a target serum uric acid level of <6 mg/dL. In patients with renal impairment, the starting dose is 25 to 50 mg/day.
Febuxostat. Febuxostat is a nonpurine selective inhibitor of xanthine oxidase, approved by the US Food and Drug Administration (FDA) in February 2009. It is a potent inhibitor of the enzyme that leads to significant urate reduction.17,21,22
Febuxostat might have an advantage over allopurinol with its potential role in patients who are allergic or refractory to allopurinol therapy if there are no other contraindications.
Results are comparable with allopurinol. In a study comparing febuxostat with allopurinol in patients with gout and urate levels of at least 8 mg/dL, urate levels dropped below 6 mg/dL in 53% of patients who received 80 mg of febuxostat, whereas this level was achieved in only 21% in the allopurinol group.17 The response was greater in the group taking 120 mg of febuxostat. This response was also consistent with reduction in tophus size in both groups.
This study showed the superiority of febuxostat to 300 mg of allopurinol and it emphasized the importance of titrating the dose to reach target levels instead of using a standard dose of 300 mg in all patients. Furthermore, after cessation of prophylaxis (colchicine or naproxen was used) 8 weeks after starting febuxostat or allopurinol, the incidence of gout flares doubled in the group taking 120 mg of febuxostat and tripled in the group taking 80 mg of febuxostat and 300 mg of allopurinol. This result supports the concept of necessary long-term prophylaxis for acute gout attacks after hypouricemic therapy is initiated.
Other studies do not necessarily indicate that febuxostat is superior to allopurinol.19,23
Febuxostat is well tolerated without the need for dosage adjustment even in patients with mild renal impairment. The most frequently reported adverse effects are diarrhea, back pain, headaches, and arthralgias. Also, patients should be monitored for thromboembolic events and increased hepatic transaminases. Adverse effects were not increased in patients with moderate renal insufficiency (creatinine 1.6–2.0 mg/dL). Doses used in trials were 40, 80, and 120 mg/day orally; however, 80 mg may be the best dose in clinical practice. The doses should be based on the patient's serum urate levels, as previously discussed.
Similar to allopurinol, febuxostat carries drug interactions with azathioprine and mercaptopurine by sharing the enzyme xanthine oxidase. See allopurinol section for cautions and contraindications for use.
Uricosuric Drugs. One uricosuric agent is FDA approved as monotherapy: probenecid. Lesinurad is also in this class but is approved only in combination with a xanthine-oxidase inhibitor. Benzbromarone, which has shown some efficacy, is not available in the US.
This drug class inhibits the urate-anion exchanger in the proximal tubule that mediates urate reabsorption, thus leading to increased urate excretion through the kidneys. Patients need to maintain good urine volume and adequate hydration when taking these agents.
Before starting a uricosuric agent, the 24-hour urine excretion of uric acid should be checked. These drugs are to be avoided in patients who have overproduction of urate or a history of urate nephrolithiasis.
It is advisable to start probenecid at a low dose and increase the dose gradually until reaching the target serum urate level. The starting dose is 250 mg twice a day. The maintenance dose varies, averaging 500 to 1,000 mg 2 or 3 times a day. The maximum effective dose is 3,000 mg/day.
Adverse effects include rash, precipitation of acute gouty arthritis, GI intolerance, and uric acid stones. Probenecid increases urinary calcium excretion in patients with gout and, thus, is contraindicated in patients with a history of calcium or urate nephrolithiasis. Drug interactions with probenecid include interference with penicillin and ampicillin excretion and autoimmune hemolytic anemia.
Uricase. The active ingredient in this drug is uricase, the enzyme that catalyzes conversion of urate to allantoin. Humans lack this enzyme, making them unable to catabolize urate naturally. The enzyme itself is highly immunogenic. Uricase can lower urate levels, resulting in gout flare reduction and tophus resolution. Two drugs are available: Pegloticase and rasburicase, but only pegloticase is approved for gout. Rasburicase use is limited to preventing acute uric acid nephropathy in patients with leukemia, lymphoma, or solid tumor malignancies receiving chemotherapy.
Pegloticase is a recombinant pegylated form of uricase, the enzyme that catalyzes conversion of urate to allantoin. It has been shown effective in treating chronic gout in adult patients refractory to conventional therapy. Labeling recommends that pegloticase be administered at 8 mg intravenously every 2 weeks with emphasis on premedication with acetaminophen, an antihistamine, or a systemic steroid to minimize the risk of anaphylaxis and infusion reactions. Patients should be monitored for approximately 1 hour after the infusion. Prior to every infusion, serum urate testing should be obtained. If the urate levels are rising, infusions should be held as this may indicate a high risk for infusion reaction and anaphylaxis. Due to the development of immunogenicity, use is not recommended in combination with other urate-lowering agents.
Pegloticase is effective in decreasing tophi burden (Figures 12-13). Pegloticase, however, carries a higher risk for infusion reaction than other urate-lowering agents. It is not recommended to use as first-line therapy unless no other agent can be used. The ACR advises its use for refractory gout and chronic tophaceous gouty arthropathy if other urate-lowering agents have failed, were not tolerated, or are contraindicated.15
It is important to avoid high-risk medications that could lead to hyperuricemia, such as diuretics, cyclosporine, and tacrolimus as well as other medications with uricosuric action. If a patient with gout and hyperuricemia requires therapy for hypertension, losartan may be a better choice than a diuretic.
Although some food and beverages are associated with hyperuricemia and gouty attacks, dietary factors are not sufficient to explain most gout attacks. Nevertheless, patients should be encouraged to follow healthy diets and to avoid excessive consumption of alcohol (especially beer), shellfish, and purine-rich food (meat in general, especially organ meats).24,25 Patients whose diets are low in dairy products have a higher risk of developing gout attacks.25,26 Encourage patients to adequately hydrate to avoid dehydration.
Young patients. Gout or hyperuricemia in an adolescent or child is rare but invariably a manifestation of an underlying metabolic or inherited enzyme deficiency warranting a workup for these diseases.
Postmenopausal women. Gout is more common in postmenopausal women. In premenopausal women, their higher estrogen levels provide a uricosuric effect that is lost after menopause, which increases the risk of developing gout.27 Gout in postmenopausal women tends to involve the upper extremities.
Transplant recipients. Transplant recipients have an increased chance of drug-drug interaction, high-risk medication use, and risk of organ failure. Use allopurinol with caution in transplant patients taking azathioprine. Combining azathioprine and allopurinol increases azathioprine blood levels, which increases the risk of bone marrow suppression.28 Commonly used immunosuppressant drugs such as cyclosporine and tacrolimus can increase urate levels.
Metabolic syndrome. Existence of the metabolic syndrome needs to be evaluated in patients with hyperuricemia. It is essential to treat conditions causing the metabolic syndrome, such as hypertension, overweight, dyslipidemia, and insulin resistance, in addition to treating gout. When an antihypertensive agent is being considered, losartan offers an advantage because it has mild hypouricemic properties. Prevention of obesity and hypertension might decrease the incidence of gout and morbidity.
Calcium pyrophosphate deposition (CPPD) disease is a crystal deposition disease in the joints and soft tissue, resulting in inflammation and tissue damage. The clinical presentation resembles gout in its acute attacks of crystal synovitis and, thus, was previously called pseudogout. Acute CPPD arthritis is now the preferred term for this disease.
If a radiographic joint examination shows calcification of cartilage, the syndrome is called chondrocalcinosis. Although CPPD crystal deposition and chondrocalcinosis are seen in acute CPPD arthritis, not all patients with either chondrocalcinosis or CPPD crystal deposition present with acute arthritis.
Few studies have addressed the epidemiology of CPPD disease. It appears to affect about 1 per 1000 individuals and is much more common in those older than 65 years of age. A widely cited study that looked at age distribution of patients with CPPD disease by radiographic examination reported the prevalence to be 15% in those aged 65 to 74 years and higher than 40% after age 84.29 This disease is not commonly encountered in younger patients (ie, under 50 years of age) without a previous history of trauma or surgery. It has no definite ethnic or gender predilection.30
Epidemiologic studies, however, have not been consistent in using universal investigative methods. Some studies estimated the prevalence based on radiographic findings of chondrocalcinosis in degenerative joint diseases; other studies estimated it based on synovial fluid analyses. This discrepancy creates limitations in extrapolating the data to patients with the clinical constellation of CPPD disease symptoms.
Genetic factors are believed to cause familial autosomal dominant CPPD chondrocalcinosis. Studies are investigating mutations of the ANKH gene and chondrocalcinosis but the role of gene mutations is not fully understood.
Although the pathogenesis of CPPD disease is not as well understood as the pathogenesis of gout, there is probably excessive pyrophosphate production in cartilage resulting in calcium pyrophosphate super saturation and the formation of CPPD crystals.31,32
Chondrocalcinosis and CPPD crystals may be associated with certain underlying diseases such as trauma to the joint, hyperparathyroidism, hypomagnesemia, hypophosphatasia, hypothyroidism, and hemochromatosis. This highlights the importance of addressing other possible underlying diseases when evaluating the patient with CPPD and chondrocalcinosis.
The CPPD disease encompasses a variety of clinical manifestations:
Calcium pyrophosphate deposition disease is distinguished by acute attacks of synovitis that mimic gout. These acute or subacute attacks can involve one or multiple joints, but usually not more than four or five joints.33 Similar to gout, CPPD disease can manifest with systemic features such as fevers, malaise, leukocytosis, and elevated acute-phase reactants (sedimentation rate and CRP). Acute attacks may be indistinguishable from acute gout. In fact, it is often difficult to differentiate both without a synovial fluid analysis. Patients have joint pain, synovitis with joint tenderness, and swelling. Although CPPD disease and gout share similar joint predilection, CPPD disease tends to affect larger joints (knee joints) more commonly than gout and smaller joints (first metatarsophalangeal joints) less commonly than gout. Calcium pyrophosphate deposition disease also affects the elbow, shoulder, wrist, and metacarpophalangeal joints.
Because acute CPPD disease closely resembles gout, the definitive diagnosis often requires synovial fluid analysis. Synovial fluid should be microscopically analyzed for cell count and crystal analysis under compensated polarizing microscopy. In addition, fluid should be examined by Gram stain and culture, especially if crystals are not found. On synovial fluid polarization, CPPD crystals might not be as evident as MSU crystals. They are weakly birefringent under polarized light and have a rhomboid or rod-shaped appearance. They are seen either intracellularly or extracellularly; however, detection might not be as accurate if fluid analysis is delayed. In addition, because CPPD disease and gout can coexist, MSU crystals might be observed. White cell counts can range from a few thousand cells up to 80,000 to 100,000 per high-power field.
Radiographs can show chondrocalcinosis in the joint involved and other more typical joints even if CPPD disease is not clinically active at the time of presentation. Radiographs can help confirm the clinical impression (especially the knee joints, wrists, and anteroposterior view of the pelvis) and extent of joint degeneration; however, radiographs are not required to make the diagnosis once CPPD crystals are seen under polarized light. Chondrocalcinosis is seen in the knees (hyaline cartilage and menisci), the wrists (fibrocartilage), and other joints such as intervertebral discs and symphysis pubis. Other radiographic features include joint space narrowing, subchondral new bone formation, normal mineralization, cysts more prominent than in osteoarthritis, bilateral preponderance, and osteophyte formation. Certain metabolic conditions associated with CPPD disease, such as in hemochromatosis, have characteristic findings such as joint narrowing of the metacarpophalangeal joint spaces, squaring of the bone ends, subchondral cysts, and hook-like osteophytes on the radial aspects of the metacarpal heads, especially the second and third metacarpophalangeal joints (Figures 14-15).34
Most of the differential diagnosis factors with gout can be considered here. Infection is always a major differential, especially in the patient presenting with new acute monoarticular arthritis. In addition, septic arthritis can coexist in a joint that has been or is involved in an acute CPPD disease attack as with gout. Thus, it is important to aspirate the involved joint whenever possible for microscopic examination of the synovial fluid and Gram stain and culture. Other differentials include trauma, bleeding, and other crystal-deposit diseases. Calcium pyrophosphate deposition disease may mimic polymyalgia rheumatica.
Optimal therapy promptly treats an acute attack, prevents additional attacks, and prevents or reverses the degenerative joint disease associated with CPPD disease arthropathy. Unfortunately, no proven therapy fits this description.
The treatment of CPPD disease is mostly tailored to the manifesting symptoms. In patients presenting with one or two points of acute synovitis and no infection, rapid relief of pain and inflammation is accomplished with joint aspiration and steroid injection. Many patients find relief from the joint aspiration itself. When more than two joints are involved, it is not feasible to inject all the joints, so treatment is directed more toward systemic therapy.
The side effects and toxicities of NSAIDs, colchicine, or systemic glucocorticoids are similar to those for patients with gout.
Colchicine at a dose of 0.6 mg once or twice daily may be effective as a prophylactic measure to reduce the number of attacks in a year, especially in patients who experience three or more attacks a year.35 Unlike gout, however, there are no hypouricemic equivalents to improve the long-term control of acute attacks or to prevent or reverse CPPD disease.
The outcome of patients with CPPD disease is influenced by genetic predisposition, extent of crystal deposition and joint degeneration, and aggravating factors from the underlying associated diseases. A study that followed 104 patients with pyrophosphate arthropathy for a mean of 4.6 years found that patients presenting with acute attacks have a good prognosis.36 They also found that some patients did not have progressive disease.
Because CPPD disease is associated with variety of underlying conditions, practitioners should screen for hyperparathyroidism, hypothyroidism, hypomagnesemia, hypophosphatasia, and hemochromatosis. Blood should be tested for intact parathyroid hormone, calcium, phosphorous, thyroid-stimulating hormone, magnesium, ferritin, iron transferrin, and alkaline phosphatase. Treatment is recommended for any associated diseases; however, it is unclear if treatment of comorbid conditions would decrease the chondrocalcinosis or reverse joint degeneration.
Crystal deposition disease is a relatively common condition. Gout and CPPD disease are the most common of these disorders, but practitioners need to be aware of the presence of other types of crystal arthropathy, such as hydroxyapatite crystal deposition disease. In this case, crystals might not be seen on classic synovial analysis and some may require special staining.
Gout and pseudogout can manifest with similar symptoms, and their clinical presentation might not be distinguishable; thus, it is essential to aspirate the affected joint or bursa for synovial fluid and crystal analysis whenever possible. Cell count, Gram stain, and culture, in the right clinical setting, should be sought.
Once the diagnosis is made, treatment for acute attack should be commenced using the least toxic agent or the one that carries least risk for the patient. Treatment should be initiated while taking into consideration other comorbid conditions, such as renal disease, gastric disease, organ transplant, drug interactions, and others, because these will affect the choice of therapy. In the case of gout, once the acute disease has resolved, the patient should be followed to assess for indications and need for hypouricemic therapy.
New drugs are on the horizon for managing chronic tophaceous gout. Over the years, several medications have been approved for the management of gout, such as febuxostat and pegloticase. Additional medications, such as IL-1 inhibitors and biologic therapy are being investigated, allowing for additional treatment options for acute crystalline arthritis with gout and CPPD disease. It is essential for practitioners to keep current with new research data on the indications, safety, and efficacy of these drugs to use them competently in clinical practice.
Feyrouz Al-Ashkar, MD; nothing to disclose.