Published: December 2013
Last reviewed: June 2017
Autoimmune mucocutaneous blistering diseases (AMBDs) are a group of conditions that manifest with blisters on the skin or mucous membranes. In order to get the most accurate diagnosis when performing biopsies in AMBD, the tissue for hematoxylin and eosin (H&E) should be taken from the edge of a blister and the sample for direct immunofluorescence (DIF) should be taken from perilesional skin.
Bullous pemphigoid (BP) is the most common AMBD with an estimated prevalence of 1 in 40,000. It usually manifests after the 6th decade of life, and there is a slightly higher incidence in women than in men. Rarely, children are affected.
Triggering factors in BP can be identified in up to 15% of patients. Medications, trauma (surgical procedures, radiation, burns, vaccines), and infections can all induce BP. The cutaneous manifestations vary. Typically, there is a prodrome, or non-bullous phase, in which patients have pruritus and urticarial or eczematous plaques on the trunk and extremities that can last for weeks to months.
Acral sites such as the palms can also be involved. During the non-bullous phase, the condition is often misdiagnosed as urticaria.
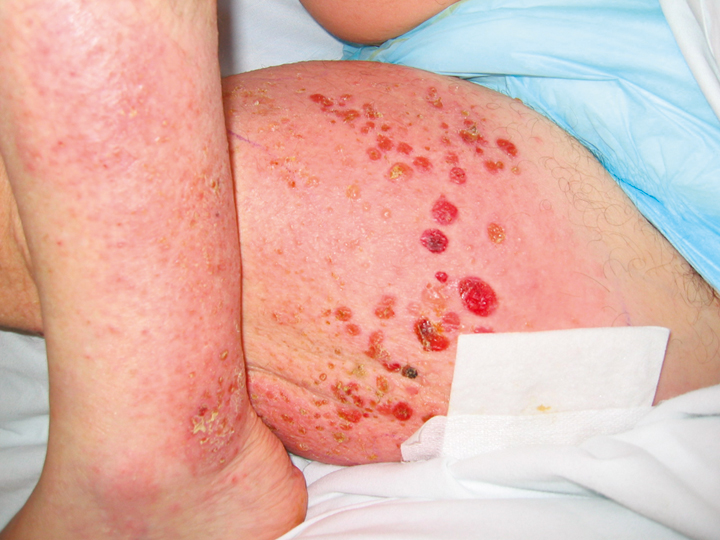
During the bullous phase, tense vesicles and bullae develop on normal or erythematous skin (Figure 1). Blisters heal leaving erosions, crust, and dyspigmentation. Bullae can reach a size of several centimeters before rupturing. Blisters usually appear in a symmetric distribution; however, flexures and the abdomen are the most common sites. Blisters in the oral cavity occur in up to 30% of patients, and they usually occur when the cutaneous disease is extensive. Occasionally, BP can be induced by systemic medications such as diuretics (furosemide), analgesics (NSAIDs), antihypertensive agents (captopril), and antibiotics (amoxicillin, ciprofloxacin, penicillin). Pemphigoid gestationis is a variant of BP that occurs during pregnancy.
Skin biopsies for H&E staining and DIF confirm the diagnosis. Histologic findings of a blister show a subepidermal bullae with an inflammatory infiltrate of predominantly eosinophils with neutrophils and histiocytes in the upper dermis. Perilesional, uninvolved skin is ideal for DIF. DIF demonstrates fine, linear IgG and C3 at the basement membrane zone. Biopsies for DIF should be placed in Michel's transport media for processing rather than saline to avoid false-negative results.
Serologic studies for indirect immunofluorescence (IIF) can help to confirm the diagnosis. Circulating IgG antibodies to the basement membrane zone can be detected in approximately 80% of patients with BP. Additionally, a peripheral blood eosinophilia may be present, occurring in half of BP patients. Diagnosis of BP is summarized in Box 1.
| Bullous Pemphigoid |
|---|
| H&E: Subepidermal bullae |
| DIF: Linear C3 and IgG at basement membrane zone |
| Pemphigus Vulgaris |
| H&E: Intraepidermal acantholysis |
| DIF: Intercellular IgG and C3 in the epidermis |
| Mucous Membrane Pemphigoid |
| H&E: Subepidermal bullae |
| DIF: linear IgG and C3 deposition at the basement membrane zone |
| Pemphigus Foliaceus |
| H&E: Superficial epidermal acantholysis |
| DIF: Intercellular IgG deposition in the stratum corneum |
DIF, direct immunofluorescence; H&E, hematoxylin and eosin; Ig, immunoglobulin.
Systemic corticosteroids provide the most rapid and effective control of disease. Initial doses of oral prednisone at 0.5 to 1.0 mg/kg per day, based on ideal body weight, often halts formation of new blisters. The dose of prednisone can be tapered slowly over a period of several months to a maintenance dose between 5 and 10 mg/day. It is essential to start these patients on oral calcium and vitamin D supplements, as many of them will remain on prednisone for months.
When the dose of prednisone cannot be lowered to less than 10 mg/day due to continued blister formation, immunosuppressive agents (ISAs) such as methotrexate, mycophenalate mofetil, dapsone, azathioprine, or cyclophosphamide may be added. In cases refractory to this conventional immunosuppressive therapy, intravenous immunoglobulin (IVIg) administered monthly has been shown to induce control of disease, and continued use following a defined protocol has led to long-term remissions. Rituximab has been used successfully in refractory cases.
It is essential that frequent bacterial cultures of cutaneous erosions be performed to identify early infections. Erosions may become secondarily infected, especially when patients are taking ISAs for treatment of BP. Appropriate antibiotics should be initiated promptly. To aid in healing, moist cutaneous erosions, topical soaks with aluminum acetate (Domeboro) for 10 minutes, two to four times a day, is very beneficial. Treatment of BP is summarized in Box 2.
| Bullous Pemphigoid |
|---|
| Prednisone and dapsone, taper off prednisone to <10 mg/day |
| If refractory to prednisone <10 mg/day, add an immunosuppressive agent |
| If refractory to conventional therapy, try IVIg or rituximab |
| Pemphigus Vulgaris |
| Prednisone plus an immunosuppressive agent |
| IVIg or rituximab in refractory cases |
| Mucous Membrane Pemphigoid |
| Topical and intralesional corticosteroids |
| Dapsone ± prednisone |
| Prednisone plus an immunosuppressive agent, if unresponsive |
| Pemphigus Foliaceus |
| High-potency topical corticosteroids |
| Hydroxychloroquine |
| Prednisone plus an immunosuppressive agent |
| IVIg or rituximab in refractory patients |
IVIg, intravenous immunoglobulin.
Pemphigus vulgaris (PV) is a chronic, intraepidermal AMBD in which autoantibodies bind to desmogleins which are cell-adhesion proteins. It typically manifests in the 5th or 6th decade of life. It occurs in both sexes with equal frequency. It can occur in all ethnic groups but is most common in those of Ashkenazi Jewish descent. The prevalence in Jerusalem is 16 cases per million, compared to 1 case per million in the rest of the population.
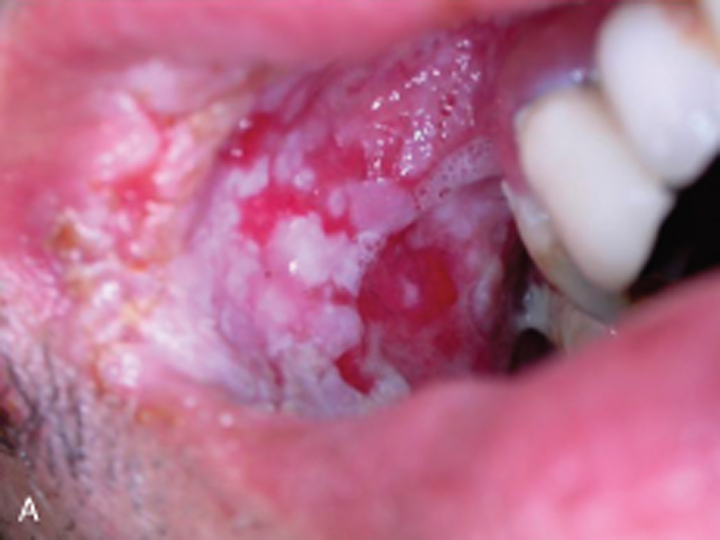
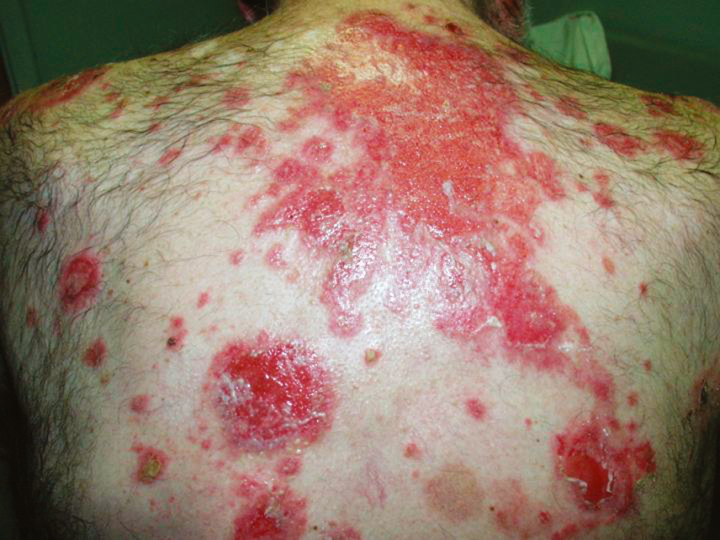
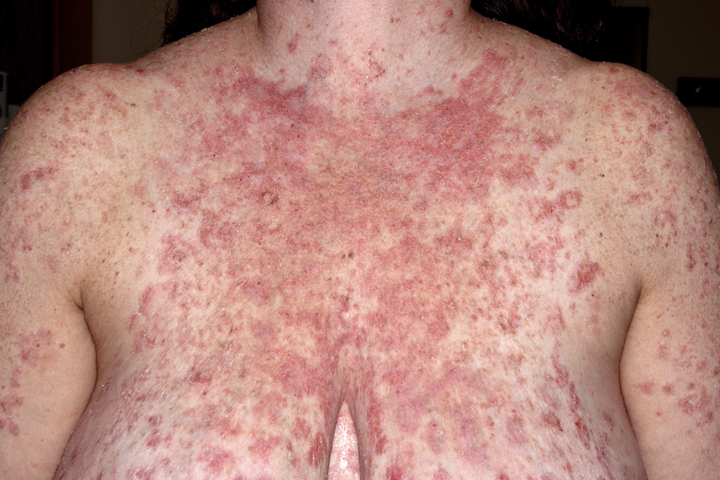
Patients present with painful erosions in the oral mucosa. The earliest erosions are usually located on the buccal mucosa, adjacent to the lower molars. However, the fixed gingiva, soft palate, and tongue are often affected as well. It is uncommon to see an intact blister on mucosa. Rather there are erosions with a characteristic jagged appearance (Figure 2). Large and painful erosions may result in decreased oral intake. When the disease is limited to the oral cavity, it is often misdiagnosed as an infection, including candidiasis or herpes. Half of PV patients will also present with cutaneous erosions. Blisters quickly rupture and become crusted erosions that may not heal. Erythema surrounding the base of a blister is variable. Common sites of involvement are on the scalp, face, chest, upper arms, and back (Figure 3).
Any mucosal site can be affected. Nasal involvement manifests with bleeding and crusting. The upper one third of the esophagus can be affected, resulting in dysphagia. A hoarse voice may indicate vocal cord involvement. Erosions can also occur on the palpebral conjunctiva and the genital and anal mucosa (Boxes 1 and 2).
Oral or cutaneous biopsies for H&E staining and DIF are needed to confirm the diagnosis. Histopathologic findings of an early vesicle show intraepidermal acantholysis, which is a loss of adhesion between keratinocytes. Normal skin adjacent to an early blister is ideal for DIF, which shows intercellular IgG and C3 within the epidermis.
Serologic studies for IIF should be performed at the same time as the biopsies. Circulating intercellular IgG antibodies can be detected in approximately 80% of patients with PV. A more sensitive enzyme-linked immunosorbent assay (ELISA) is available for detecting antibodies to desmogleins 1 and 3, intercellular adhesion proteins. Diagnosis of PV is summarized in Box 1.
PV is typically more difficult to treat than BP. The circulating autoantibody binds directly to the intercellular proteins that hold keratinocytes together, thereby causing acantholysis. Neither complement fixation nor an inflammatory response is required for blister formation. Thus, medications with anti-inflammatory effects that can benefit patients with BP are not as effective in patients with PV. Suppression of autoantibody formation is important for long-term control of disease.
The cornerstone of management for PV includes both corticosteroids and other ISAs. Systemic and topical corticosteroids provide the most rapid and effective control of disease. Prednisone at a dose of 0.5 to 1.0 mg/kg per day, based on ideal body weight, often halts formation of new blisters. Prolonged treatment of PV can result in high cumulative doses of corticosteroids, which are the main causes of treatment-related side effects. Often, ISAs such as methotrexate, mycophenolate mofetil, or azathioprine are used together with prednisone at the onset of treatment. The long-term goal is to taper the dose of prednisone slowly over a period of several months to a maintenance dose between 5 and 10 mg/day. IVIg, administered in monthly cycles following a defined protocol, and rituximab have shown promise in treating patients with refractory disease.
Obtaining frequent bacterial cultures of open cutaneous erosions and topical therapies as discussed for BP greatly enhances the success of the systemic therapy.
To minimize the formation of new oral blisters, patients should avoid eating foods with jagged edges such as potato chips, toast, and pretzels. A soft toothbrush should be used and care should be taken not to aggressively brush the fixed gingiva. The regular use of a steam inhaler, particularly in months of low humidity, can greatly decrease the amount of nasal crusting and bleeding. Treatment of PV is summarized in Box 2.
Mucous membrane pemphigoid (MMP), also termed cicatricial pemphigoid, is a subepidermal blistering disease that typically manifests in the 6th or 7th decade of life. The female-to-male ratio is about 2:1.
MMP differs from BP in that individual lesions heal with scarring. There are varying manifestations of the disease depending on which protein in the basement membrane is involved. A common presentation is that of smooth-bordered erosions in the oral mucosa. The most common location is the oral mucosa, with approximately 90% of patients having oral erosions. If the oral mucosa is the only mucosal site involved, the condition is termed oral pemphigoid.
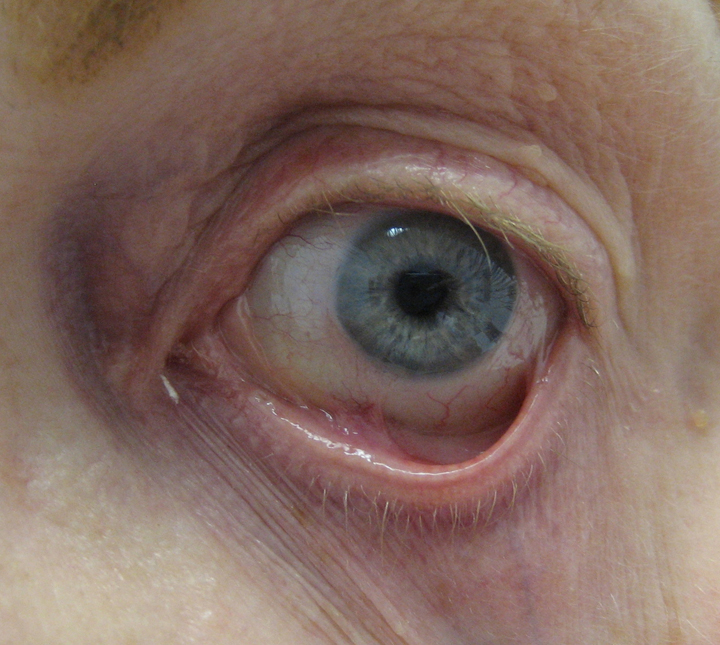
As in PV, any mucosal site can be affected. Involvement of the nasal mucosa can lead to strictures and can be associated with lesions of the upper aerodigestive tract. Rarely, airway obstruction can occur. Repeated coughing and a hoarse voice may be indicators of blistering in the pharynx and larynx. Esophageal involvement can present as dysphagia, but can also be asymptomatic. Repeated blistering and subsequent scarring in these areas can result in strictures. Involvement of the conjunctiva, which manifests clinically as conjunctivitis and xerosis, results in scarring between the palpebral and bulbar conjunctiva (symblepharon), entropion, and subsequent trichiasis. Progressive scarring can lead to blindness. An example of symblepharon is shown in Figure 4.
Involvement of the genital and anal mucosa is fairly common. Persistent blistering and scarring of the vaginal mucosa can result in stenosis that prohibits screening pelvic examinations. Anal involvement manifests as localized pain and bleeding, which can lead to stenosis if left untreated.
Cutaneous tense blisters similar to those seen in BP occur in only 25% of patients. The scalp, face, neck and upper trunk are the most frequently involved sites. Healing occurs with pink, atrophic scarring.
Another type of MMP, termed antiepiligrin cicatricial pemphigoid, occurs when antibodies are formed against laminins 5 in the basement membrane. In this subset, there is a six-fold increased risk of solid organ malignancies (usually adenocarcinoma). These patients with anti-laminin 5 autoantibodies cannot be distinguished clinically from other variants of MMP.
Biopsies for H&E and DIF should be performed to confirm the diagnosis. Histologically, findings are almost identical to BP. A subepidermal vesicle is seen with an inflammatory infiltrate of neutrophils and eosinophils in the upper dermis. Scarring can be seen in the upper dermis. DIF of perilesional mucosa reveals linear IgG and C3 deposition at the basement membrane zone in 95% of patients. Because there are low amounts of circulating antibodies, IIF testing is not generally helpful. Diagnosis of MMP is summarized in Box 1.
Patients with localized oral involvement often respond to clobetasol gel or intralesional triamcinolone injected every 4 to 6 weeks as needed.
Patients with multiple mucosal sites should be treated with systemic therapy including ISAs such as methotrexate, mycophenolate mofetil, dapsone, azathioprine, or cyclophosphamide Patients with eye disease require aggressive treatment to prevent blindness; the treatment of choice is cyclophosphamide. Treatment of MMP is summarized in Box 2.
Pemphigus foliaceus is an AMBD that typically manifests in the 6th or 7th decade of life. Fogo selvagem, the endemic form of pemphigus foliaceus seen in Brazil and other regions of South America, commonly manifests in the 3rd and 4th decades. It is transmitted by the Similium black fly. In Western Europe, the incidence of pemphigus foliaceus is 0.5 to 1 case per million population per year. In Brazil, where the endemic form of pemphigus foliaceus is common, the incidence is as high as 50 cases per million population per year.
Pemphigus foliaceus is characterized by superficial flaccid bullae that easily rupture, leaving erosions. Blisters appear on an erythematous base with associated crusting and scaling. Lesions can be mistaken for impetigo. Patients typically present with crusted erosions on the scalp, face, and upper trunk in a seborrheic distribution (Figure 5). The disease may stay localized; rarely it can progress to generalized involvement and erythroderma. In contrast to PV, pemphigus foliaceus does not involve the oral mucosa.
Pemphigus foliaceus can be idiopathic or it may be triggered by certain medications such as penicillamine, captopril, or other thiol drugs. Pemphigus foliaceus occurs in up to 7% of patients treated with penicillamine, and it can occur up to 1 year after starting treatment. An arthropod vector carrying an infectious agent can induce fogo selvagem in endemic areas.
Biopsy of lesional and perilesional skin is needed to confirm the diagnosis. Histopathology shows acantholysis in the superficial layers of the epidermis. DIF reveals intercellular IgG deposition in the superficial epidermis. The antibody is of the IgG4 subclass and directed against desmoglein 1 (dsg-1).
Serum can be tested for IIF and ELISA to detect anti–dsg-1 antibodies. Pemphigus foliaceus can be distinguished from PV, because patients with pemphigus foliaceus only have antibodies directed against dsg-1 (not desmoglein 3). Diagnosis of pemphigus foliaceus is summarized in Box 1.
All patients should be encouraged to avoid sun exposure because it can exacerbate the condition. Any trauma or pressure to the skin commonly produces new lesions.
High-potency topical corticosteroids can be used for mild cases of pemphigus foliaceus. In more-extensive cases, suppression of autoantibody formation with systemic corticosteroids and ISAs is considered conventional therapy. Prednisone at a dose of 0.5 to 1.0 mg/kg per day, based on ideal body weight, often halts formation of new blisters. Hydroxychloroquine has been shown effective in some patients with pemphigus foliaceus. Another effective steroid-sparing regimen is a combination of nicotinamide 1.5 g/day and either tetracycline 2 g/day or minocycline 100 mg/day. In patients with disease refractory to these treatments, IVIg or rituximab may be effective.
Frequent bacterial cultures of open cutaneous erosions should be performed to identify early infections. Appropriate oral antibiotics should be initiated promptly. Treatment of pemphigus foliaceus is summarized in Box 2.