Published: April 2016
The myeloproliferative neoplasms (MPNs), previously termed the myeloproliferative disorders, are characterized by the clonal proliferation of one or more hematopoietic cell lineages, predominantly in the bone marrow, but sometimes in the liver and spleen.1 In contrast to myelodysplastic syndromes (MDS), MPNs demonstrate terminal myeloid cell expansion into the peripheral blood. In the 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms, MPNs include chronic myelogenous leukemia (CML), chronic neutrophilic leukemia, polycythemia vera (PV), primary myelofibrosis (PMF), essential thrombocythemia (ET), chronic eosinophilic leukemia, mastocytosis, and unclassifiable MPNs.2 MDS/MPN overlap disorders are those chronic myeloid disorders unable to be classified as "classic" MPN or MDS. These include chronic myelomonocytic leukemia, atypical CML, juvenile myelomonocytic leukemia, and unclassifiable MDS/MPN.
Chronic myelogenous leukemia is the only MPN that is characterized by the presence of the BCR-ABL fusion gene, which is formed by translocation of the ABL gene from chromosome 9 joining to the BCR gene on chromosome 22. The altered chromosome 22 with the fusion gene is the Philadelphia chromosome. With a unique pathogenesis and treatment, CML is often considered separately from the rest of the MPNs. The most commonly recognized mutation in the remainder of the Philadelphia chromosome-negative MPNs is Janus kinase 2 (JAK2) V617F, which is present in more than 90% of patients with PV and approximately half of those with PMF or ET (Table 1).3 This mutation substitutes phenylalanine for valine at position 617 in the JH2 domain (Val617Phe, V617F) of exon 14, leading to constitutive activation of the JAK-STAT and other pathways resulting in uncontrolled cell growth.4 Subsequent to this, additional mutations within exon 12 of JAK2 have been identified in PV.5 Mutant JAK2 allele burden might be important in identifying high-risk patients with PV or ET (ie, those at risk for requiring treatment with chemotherapy or those at risk for developing major cardiovascular complications).6
Mutations within the thrombopoietin receptor gene (MPL) also have been identified in ET and PMF.7 The presence of these mutations is determined by polymerase chain reaction (PCR) assays and may be helpful in differentiating a MPN from a reactive cause for elevated counts. More recently, mutations in the gene that encodes calreticulin (CALR), have been identified in a large proportion of patient with MPN who did not have a JAK2 or MPL mutation.8,9 These three "driver mutations" are often mutually exclusive, meaning that if one is present the others are absent. Nonetheless, roughly 10% of patients with ET or PMF lack JAK2, CALR, or MPL gene mutations and have been referred to as being "triple-negative."
This chapter reviews the definition, epidemiology, pathophysiology, signs and symptoms, diagnosis, treatment, and outcomes of the Philadelphia chromosome-negative MPNs—PV, PMF, and ET. Chronic myelogenous leukemia and chronic myelomonocytic leukemia are discussed in the Chronic Leukemia section.
| Gene | Polycythemia Vera | Essential Thrombocythemia | Primary Myelofibrosis |
|---|---|---|---|
| JAK2 | 97% | 50 - 60% | 50 - 60% |
| CALR | 3 - 10% | 3 - 10% | |
| MPL | 33% | 33% | |
| Unmutated JAK2, CALR, MPL | 10 - 15% | 10 - 15% |
Data from Nangalia J, Green TR. The evolving genomic landscape of myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program 2014; 2014:287–296.
Polycythemia vera is a clonal disorder characterized by the overproduction of mature red blood cells. Myeloid and megakaryocytic elements are also often increased. No obvious cause exists. Genetic and environmental factors have been implicated in rare cases. Familial PV has been associated with mutation of the erythropoietin (EPO) receptor.10 An increased number of cases were reported in survivors of the atomic bomb explosion in Hiroshima during World War II.
Polycythemia vera typically occurs in the 6th or 7th decade of life and occurs more commonly in men and in both men and women of East European Jewish ancestry.5,11 In the United States, the estimated prevalence of PV ranges from 44 to 57 cases per 100,000 people.12
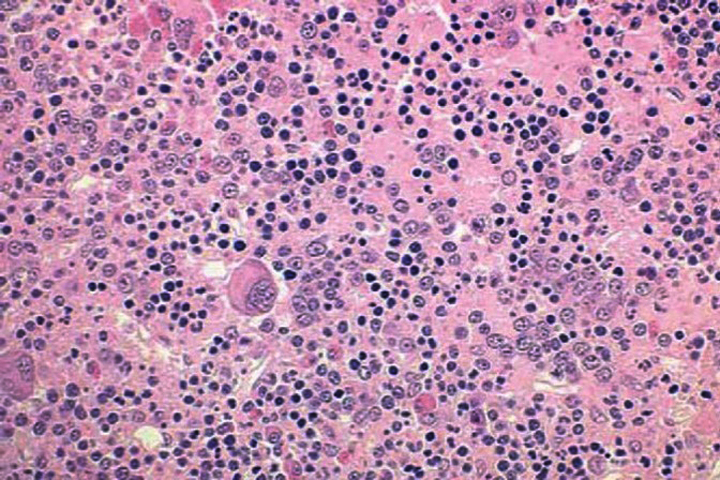
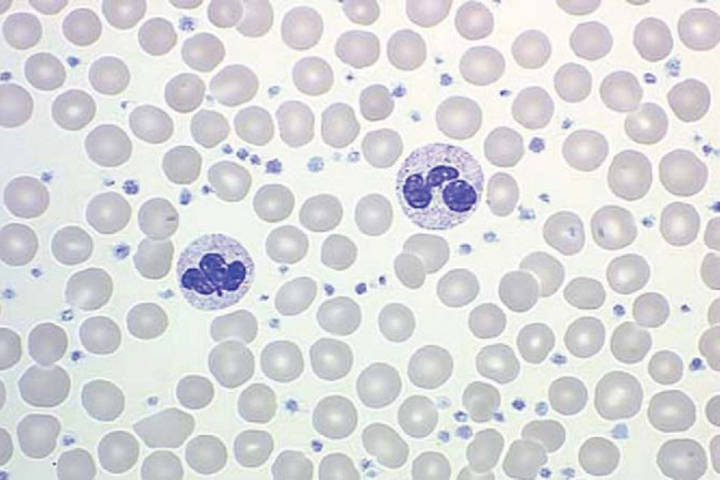
The primary defect involves a pluripotent stem cell capable of differentiating into red blood cells, granulocytes, and platelets.13 Clonality has been demonstrated through glucose-6-phosphate dehydrogenase studies as well as restriction fragment length polymorphism of the active X chromosome.10 Erythroid precursors in PV are exquisitely sensitive to erythropoietin, which leads to increased red blood cell production. Precursors in PV also are more responsive to cytokines such as interleukin-3 (IL-3), granulocyte-macrophage colony-stimulating factor, and steel factor. Myeloid and megakaryocytic elements are often increased in the bone marrow (Figure 1). The abnormal proliferation of PV is due to constitutive activation of the JAK-STAT pathway, with the majority of patients (>95%) harboring the V617F mutation.2 A similar JAK2 exon 12 mutation is found in the few patients lacking the V617F mutation.2
Increased red blood cell production in PV leads to an increased red cell mass and increased blood viscosity. This, in turn, can lead to arterial or venous thrombosis, bleeding, or both.11 Hematocrit is directly proportional to the number of thrombotic events. Investigators have demonstrated a reduction in cerebral blood flow in patients with hematocrits between 53% and 62%.10 An increased platelet count also can contribute to bleeding and thrombosis. Although platelet aggregation abnormalities exist in most patients, these abnormalities do not appear to correlate with the risk of bleeding or thrombosis. Increased production and breakdown of blood cells can lead to hyperuricemia and hypermetabolism.
Patients may be asymptomatic at the time of diagnosis and have only isolated splenomegaly, erythrocytosis, or thrombocytosis. However, most patients develop symptoms as the hematocrit and/or platelet count increase. Elevated white blood cell (WBC) counts have been found in 50% of patients.14 Elevated hematocrit has been associated with symptoms of hyperviscosity including headache, blurred vision, and plethora.
Thrombosis in small blood vessels can lead to cyanosis, erythromelalgia (painful vessel dilation in the extremities), ulceration, or gangrene in the fingers or toes. Thrombosis in larger vessels can lead to myocardial infarction, deep venous thrombosis, transient ischemic attacks, and stroke. A cerebrovascular event precedes the diagnosis in 35% of patients with PV.13 Unusual sites of thrombosis (splenic, hepatic, portal, and mesenteric) also tend to be seen more frequently in PV.
Of patients with Budd-Chiari syndrome (hepatic-inferior vena cava obstruction), 10% to 13% have coexisting PV; therefore, testing for the presence of JAK2 V617F mutation is part of the routine workup for unexplained liver thrombosis. Abnormalities in platelet function lead to bleeding complications that include epistaxis, bruising, and gastrointestinal and gingival bleeding in 5% to 10% of patients.15 Severe bleeding episodes are unusual. Hypermetabolism caused by increased blood cell turnover can lead to hyperuricemia, gout, stomach ulcers, weight loss, and kidney stones. A classic symptom is pruritus, especially after a warm bath or shower. As the disease progresses, many patients develop abdominal pain secondary to organomegaly.
Polycythemia vera should be suspected in men with hemoglobin greater than 18.5 g/dL and in women with hemoglobin greater than 16.5 g/dL.2 An elevated red cell mass, measured using direct tagging of red blood cells with chromium 51, was previously important in making the diagnosis but is rarely used in current practice. The presence of the JAK2 mutation is now a major criterion for diagnosis.2
Secondary causes of polycythemia must be ruled out. Many conditions can physiologically increase the production of EPO. Overproduction occurs in hypoxia (eg, pulmonary disease, high altitude, smoking-related carboxyhemoglobin, cyanotic cardiac disease, and methemoglobinemia), tumors (eg, kidney, brain, hepatoma, uterine fibroid, and pheochromocytoma), renal artery stenosis, and renal cysts. Other causes include androgen therapy, congenital erythrocytosis, EPO-receptor hypersensitivity, autotransfusion (blood doping), and self-injection of EPO. Serum EPO levels should be low to normal in patients with PV but high in patients with secondary polycythemia, although there may be some overlap. Molecular testing for the JAK2 V617F or other functionally similar mutation currently plays a central role in the diagnosis of PV as a way of separating neoplastic from reactive myeloid proliferations.
The diagnostic criteria for PV have undergone changes as new research discoveries are published; however, elevated red cell mass, hemoglobin, or hematocrit levels remain the cornerstones of diagnosis. The current (2008) WHO criteria require the presence of both major criteria and one minor criterion or the presence of the first major criterion together with two minor criteria to make a diagnosis of PV.2 However, the diagnostic threshold of PV is set to be lowered in the next iteration (Table 2), as the hemoglobin threshold for diagnosis will be below that of the current schema. The proposed revision to the WHO criteria requires the presence of all three major criteria, or the first two major criteria and the minor criterion to make the diagnosis of PV.16
| Current (2008) WHO criteria | Proposed Criteria |
|---|---|
| Major criteria | |
|
|
| Minor criteria | |
|
|
| Requires both major criteria and 1 minor criterion OR first major criterion and 2 minor criteria | Requires all 3 major criteria OR first 2 major criteria and the minor criterion |
aHemoglobin or hematocrit >99th percentile of method-specific reference range for age, sex, altitude of residence; or hemoglobin >17 g/dL men, >15 g/dL women if associated with a documented and sustained increase of at least 2 g/dL from a patient's baseline value that cannot be attributed to correction of iron deficiency; or elevated red cell mass >25% above mean normal predicted value.
Adapted with permission from Tefferi A, Thiele J, Vannucchi AM, Barbui T. An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia 2014; 28:1407–1413.
The management of PV focuses on reducing the risk of thrombosis and hemorrhage, as they are the major contributors to mortality in untreated patients. This is achieved primarily through decreasing the hemoglobin level, thereby reducing plasma viscosity and its attendant complications. In addition to aspirin, therapeutic options include hydroxyurea, interferon, and, most recently, JAK inhibitors. Nonetheless, phlebotomy remains the backbone of treatment.
The goal of therapy is a hematocrit no higher than 45% for men and 42% for women.17 This was first suggested on the basis of cerebral blood flow studies and eventually supported by randomized controlled trials including the Polycythemia Vera Study Group (PVSG)-01 and the subsequent CYTO-PV trial.18,19,20 The latter was a multicenter, randomized, controlled trial conducted to assess the benefit/risk profile of cytoreductive therapy with phlebotomy or hydroxyurea aimed at maintaining HCT below 45% versus maintaining HCT between 45% and 50% in 365 patients. After a median follow-up of 31 months, the primary composite endpoint of death from cardiovascular causes or major thrombotic events occurred less often in those with a hematocrit below 45% than in those in the high hematocrit group.20
In the PVSG-01 study, thrombotic events were increased, particularly in patients with a history of thrombosis, advanced age, or high phlebotomy requirement.19 Those results, combined with data from subsequent studies, ultimately led to the following thrombotic risk-adapted definition for PV treatment:
Low-risk patients may be treated with phlebotomy alone. High-risk patients should be treated with phlebotomy plus cytoreductive therapy, typically hydroxyurea in the first-line setting.13 However, interferon should be used in women of childbearing age and can be considered in patients who cannot tolerate hydroxyurea.13
Leukemogenic properties are an important consideration when choosing a cytoreductive agent. In the PVSG-01 study, there was an increased risk of leukemia in the 32P radiotherapy and chlorambucil arms (two or three times that seen in the phlebotomy arm).13 Because of this, hydroxyurea, is the most widely used myelosuppressive agent. Side effects of hydroxyurea include myelosuppression, macrocytosis, leg ulcers, increased creatinine level, and jaundice. A recent large study has demonstrated no increased incidence of leukemia in PV patients treated with hydroxyurea.22 For older, high-risk patients, 32P and busulfan can be used to help with issues of compliance and convenience, especially if the patient's life expectancy is less than 10 years; however, these are rarely used in the current era.
Myelosuppressive agents also should be used for symptomatic splenomegaly or pruritus intractable to antihistamines.13 Interferon-alfa may also be used in the place of hydroxyurea for myelosuppression, particularly in younger patients and in patients with intractable pruritus. Side effects of interferon include flu-like syndrome, fevers, neuritis, and fatigue; although the pegylated versions of interferon are better tolerated.10 Patients with PV who undergo surgery are at extremely high risk of developing postoperative complications if their erythrocytosis is not controlled before surgery.
On the heels of discovering mutations in the JAK2 gene at the root of PV and other MPNs, JAK inhibitor medications were developed. Currently, ruxolitinib is the only approved JAK inhibitor and is indicated for patients with PV who have unacceptable side effects or do not have good control of their hematocrit with hydroxyurea. In the RESPONSE study, ruxolitinib was tested in this population and found to be better than standard therapy in controlling the hematocrit, reducing the spleen volume, and improving symptoms associated with PV.23
All patients with PV and no drug contraindications or evidence of acquired von Willebrand syndrome should be treated with low-dose aspirin. The ECLAP study (European Collaboration on Low-Dose Aspirin in Polycythemia Vera) demonstrated an antithrombotic benefit for low-dose aspirin (100 mg/day) in patients already receiving treatment for PV.24 Furthermore, patients with erythromelalgia also experienced rapid relief of their symptoms after starting low-dose aspirin.13
If managed carefully, life expectancy for patients with PV approaches that of the normal population, with a median survival between 10 and 20 years with treatment. Major causes of death in untreated patients are thrombosis and hemorrhage. Fewer than 5% of patients develop acute myeloid leukemia.14,25 Approximately 10% to 15% of patients develop post-PV myelofibrosis at an average interval of 10 years from diagnosis.13 Most patients who develop myelofibrosis die within 3 years often from progressive bone marrow failure or further transformation to acute myeloid leukemia (blast-phase MPN).
Primary myelofibrosis (PMF) has been described as chronic idiopathic myelofibrosis and agnogenic myeloid metaplasia. In PMF, a clonal hematopoietic stem cell expansion in the bone marrow is accompanied by a reactive nonclonal fibroblastic proliferation and marrow fibrosis. As the bone marrow becomes fibrotic and normal hematopoiesis can no longer occur, extramedullary hematopoiesis (myeloid metaplasia) occurs in the liver and spleen.26
The incidence of PMF in 2008 to 2010 was estimated to be 1 per 100,000 population.12 The risk of developing PMF can be increased by exposure to benzene or radiation (eg, higher incidence in individuals exposed to radiation in Hiroshima). The disease is more common in Caucasians, and men and women are affected equally. The median age at diagnosis is 67 years.10 As noted earlier, patients with PV and other MPNs can develop myelofibrosis late in their disease course.
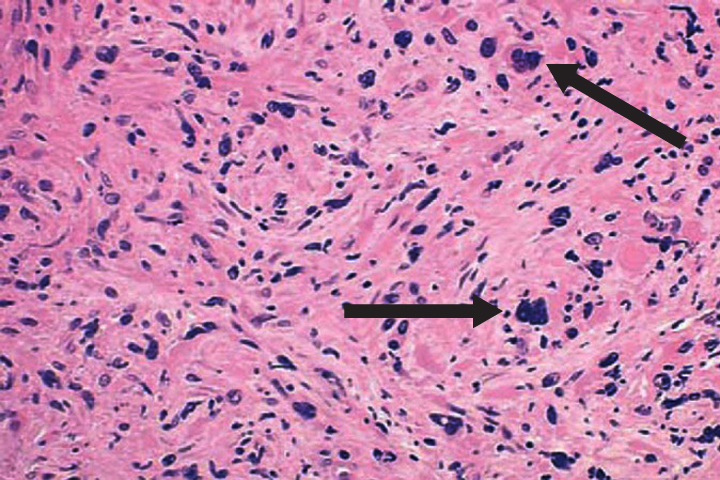
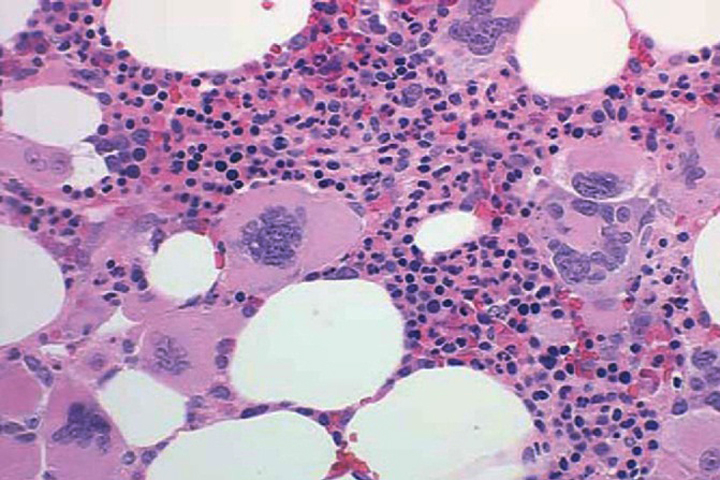
Primary myelofibrosis is described by marrow fibrosis and extramedullary hematopoiesis. Clonal studies have demonstrated a stem cell origin.26 The clonal proliferation of hematopoietic stem cells produces growth factors (platelet-derived growth factor, transforming growth factor-B, epidermal growth factor, and basic fibroblastic growth factor) leading to fibrosis of the marrow.10 Initially, the bone marrow is hypercellular, but normal hematopoiesis is diminished as the bone marrow becomes fibrotic and patients become pancytopenic (Figure 2).1 Because of this, extramedullary hematopoiesis occurs in the liver and spleen causing these organs to enlarge.
Many symptoms are attributable to the pancytopenia associated with PMF. Pancytopenia occurs as a result of decreased hematopoiesis and splenic sequestration. Most patients are anemic and feel short of breath and fatigued. Thrombocytopenia and neutropenia can lead to hemorrhage and infection, respectively.
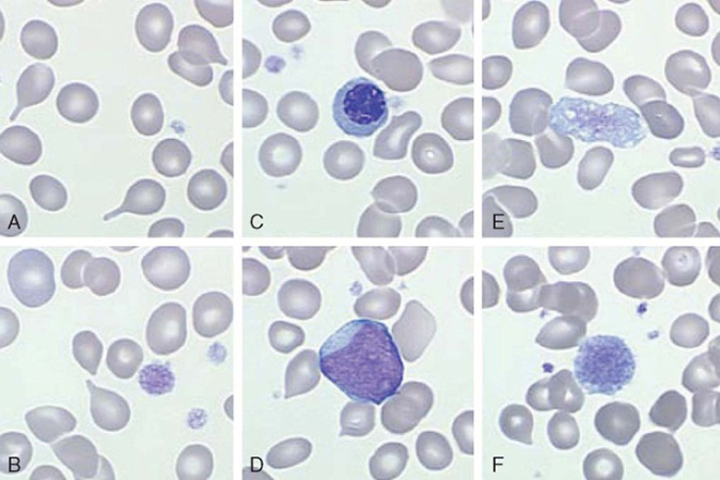
At diagnosis, up to one-fifth of patients can be asymptomatic. However, common constitutional symptoms include anorexia, weight loss, and night sweats.10 The WBC and platelet counts might increase initially but typically decrease as the disease progresses. The blood film displays a characteristic leukoerythroblastic picture (teardrop poikilocytosis, nucleated red blood cells, and immature myeloid elements) caused by crowding out of normal hematopoietic elements by fibrosis in the bone marrow (Figure 3).10
Patients might note abdominal discomfort and decreased appetite caused by splenic and hepatic enlargement. Portal hypertension and jaundice can occur as a result of increased hepatic blood flow.10 Rarely, extramedullary hematopoiesis can occur at other sites, such as the skin, lungs, bladder, genitourinary tract, gastrointestinal tract, and central nervous system. Severe bone pain typically heralds a poor prognosis and often represents a conversion of PMF to acute leukemia.
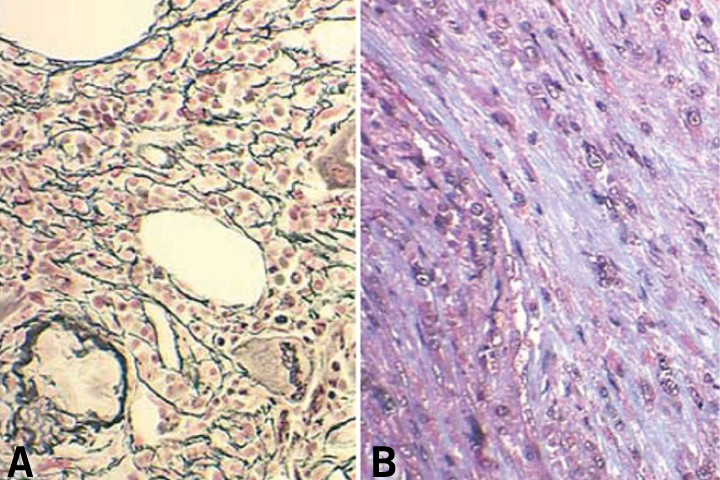
In patients with PMF, the peripheral blood smear demonstrates a characteristic leukoerythroblastic picture. Attempts to perform a bone marrow aspirate and biopsy are often complicated by a dry tap. Special stains of the bone marrow biopsy demonstrate increased fibrosis. Stains that contain silver can be used to identify reticulin, the glycoprotein coating of stromal cell strands that appears as black fibers.26 Trichrome stains identify mature collagen as bluish-green fibers, depending on the stain used (Figure 4).
The typical bone marrow biopsy is hypercellular initially, and increased numbers of abnormal megakaryocytes with a tendency to form loose to tight clusters are often present.10 Cytogenetic abnormalities are found in approximately 50% of patients.27 These abnormalities include trisomy 8, trisomy 9, del(13q), del(20q), and del(12p). Mutations in JAK2 are found in over half of patients, whereas MPL mutations are only found in approximately 5% of PMF patients. More recently, mutations in the calreticulin gene (CALR) have been described. They are found in approximately 35% of patients.3
Secondary causes of marrow fibrosis (eg, metastatic breast cancer, lymphoma, lung cancer, infection, and autoimmune disorders), as well as other hematologic disorders (eg, hairy cell leukemia, CML), need to be excluded. In addition, acute panmyelosis with MF and acute megakaryoblastic leukemia need to be ruled out. These entities also manifest with pancytopenia and marrow fibrosis, but patients typically have no splenomegaly, minimal or absent teardrop poikilocytosis, and increased numbers of blasts.
Current WHO criteria for PMF cite the following criteria for establishing a diagnosis:
Primary myelofibrosis must meet all three major criteria and two of the four minor criteria (Table 3).16
| Current (2008) WHO criteria | Proposed Criteria |
|---|---|
| Major criteria | |
|
|
| Minor criteria | |
|
|
| Requires all 3 major criteria and 2 minor criteria | Requires all 3 major criteria OR first 2 major criteria and 2 minor criterion |
aSmall to large megakaryocytes with aberrant nuclear/cytoplasmic ratio and hyperchromatic and irregularly folded nuclei and dense clustering.
bIn the absence of reticulin fibrosis, the megakaryocyte changes must be accompanied by increased marrow cellularity, granulocytic proliferation and often decreased erythropoiesis (that is, prefibrotic PMF).
cSmall-to-large megakaryocytes with aberrant nuclear/cytoplasmic ratio and hyperchromatic and irregularly folded nuclei and dense clustering.
dIn the absence of reticulin fibrosis, the megakaryocyte changes must be accompanied by increased marrow cellularity, granulocytic proliferation and often decreased erythropoiesis (that is, prefibrotic PMF).
eDegree of abnormality can be borderline or marked and institutional reference range should be used for lactate dehydrogenase level.
BM = bone marrow; CML = chronic myelogenous leukemia; ET = essential thrombocythemia; LDH = lactate dehydrogenase; MDS = myelodysplastic syndrome; PV = polycythemia vera; WHO = World Health Organization.
Adapted with permission from Tefferi A, Thiele J, Vannucchi AM, Barbui T. An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia 2014; 28:1407–1413.
The Dynamic International Prognostic Scoring System (DIPSS) includes five parameters that impact survival (Table 4).28 Total points for all five parameter are associated with four prognostic risks groups: low, intermediate-1, intermediate-2, and high risk (Table 5).28,29 Median survival decreases and incidence of blast phase to myelofibrosis increases based on an analysis of the Mayo Clinic database.30
| Parameter | Points |
|---|---|
| Age >65 years | 1 |
| Anemia: hemoglobin <10 g/dL | 2 |
| Circulating blasts: ≥1% | 1 |
| Constitutional symptoms (ie, weight loss >10%, night sweats, or fevers) | 1 |
| Leukocytosis: WBC >25 x 109/L | 1 |
Data from Passamonti F, et al. Blood 2010;115:1703–1708.
| Risk group | Points | Survival, median yrs28 | BP-MF, incidence / 100 pt-yrs30 |
|---|---|---|---|
| Low | 0 | NR | 0.3 |
| Intermediate-1 | 1-2 | 14.2 | 0.7 |
| Intermediate-2 | 3-4 | 4 | 2.6 |
| High | 5-6 | 1.5 | 8.6 |
BP-MF: blast phase to myelofibrosis.
Data from Passamonti F, et al. Blood 2010; 115:1703–1708 and Tefferi A, et al. Mayo Clin Proc 2012; 87:25–33.
The DIPSS-Plus scoring system added additional factors to further refine the risk prediction:
An unfavorable karyotype is defined as a complex karyotype or one to two abnormalities that include +8, −7/7q-, −5/5q-, inv(3), i(17q), 12p-, 11q23 rearrangement. The DIPSS remains the standard prognostic model as the data are readily available without the need for a bone marrow biopsy. Moreover, the DIPSS is dynamic, meaning that it can be calculated at any point along a patient's disease course and still hold prognostic relevance.
There are no treatments that can completely reverse the process of PMF short of allogeneic hematopoietic cell transplantation. However, because of the older median age at diagnosis, many patients are not suitable candidates for bone marrow transplant. One hematopoietic cell transplant study found a 57% relapse-free survival at 6 years in patients with myelofibrosis (median age, 51.5 yrs).32 Other studies have shown reduced regimen-related mortality (less than 10% or even 5% at day 100) and progressively improved survival with both HLA-identical sibling and unrelated donors (median age, 55 yrs).33 The increasing use of nonmyeloablative transplants should extend this option to older patients with PMF.34 Patients with intermediate-2 or high-risk PMF should be considered for bone marrow transplant.35 Patients under 60 years of age should be considered for conventional-intensity conditioning regimens, while reduced intensity (or, nonmyeloablative) transplant should be considered for those who are older. Given the limited number of FDA-approved medications, all patients should be considered for clinical trials or investigational drug therapy.
The role and benefit of using chemotherapeutic agents early in the disease to reverse the fibrosis are highly controversial, and randomized clinical trials are needed to address this issue. Therefore, care is directed toward symptomatic management with transfusions (red blood cells, platelets) and growth factors (EPO for anemia, granulocyte-colony stimulating factor for infections related to neutropenia). Androgens (eg, danazol) and, occasionally, low-dose steroids (eg, prednisone) may also be helpful in managing the anemia associated with ineffective erythropoiesis. The advent of JAK inhibitors, which are effective at reducing symptom burden, has dramatically changed the treatment landscape for primary myelofibrosis.
The immunomodulatory drugs thalidomide, lenalidomide, and pomalidomide have all been evaluated in PMF because of their anti-angiogenic, anti-TNF alpha, and T-cell modulating effects.36 Thalidomide was the first drug available. A pooled analysis of small phase 2 studies with thalidomide published between 2000 and 2002 demonstrated a response (increase in hemoglobin, or reduction or elimination of blood transfusion requirements) in 29% of patients with moderate or severe anemia.37 However, a large number of patients stopped the drug secondary to side effects (neuropathy, somnolence).
Lenalidomide has been evaluated in two separate but similarly designed phase 2 studies.38 Overall response rates were 22% for improvement of anemia, 33% for reduction of splenomegaly, and 50% for improvement of thrombocytopenia. A subset of patients had an impressive improvement in their anemia or resolution of bone marrow abnormalities (fibrosis, angiogenesis), or both. However, lenalidomide was associated with significant myelosuppression.
Pomalidomide is 20,000 times more potent than thalidomide in inhibition of TNF-alpha with a more favorable side-effect profile.39 Overall anemia response rates of 27% were demonstrated in earlier studies; however, a more recent phase 3 placebo-controlled trial found anemia response rates (16% per Delphi criteria) and response durations with pomalidomide were similar to placebo.40 The role of single-agent immunomodulatory drugs is limited, but it is being explored in combination therapy.
Splenomegaly may require treatment with myelosuppressive agents, JAK inhibitors, splenectomy, or palliative radiation. Splenic irradiation is typically associated with transient responses and should be considered for patients too ill for splenectomy or chemotherapy. Splenectomy may be considered for symptomatic splenomegaly not responding to noninvasive treatment modalities.
In a study of 223 patients at the Mayo Clinic, patients experienced durable remissions in constitutional symptoms (67%), transfusion-dependent anemia (23%), portal hypertension (50%), and severe thrombocytopenia (0%) after splenectomy.41 The surgical mortality rate was 9% and the morbidity rate was 31%, including postoperative thrombotic complications. In the patients, 16% developed hepatomegaly and 22% developed thrombocytosis, which was associated with an increased risk of perioperative thrombosis. Transformation to blast phase (acute leukemia) was 16.3%. The risk was increased in patients with splenomegaly and preoperative thrombocytopenia, suggesting that presplenectomy thrombocytopenia may be a surrogate marker of advanced disease.
Ruxolitinib, a selective JAK1/JAK2 inhibitor, is currently the only medication approved by the FDA to treat myelofibrosis. This was based on two randomized, placebo-controlled phase 3 trials: COMFORT-I and COMFORT-II.42,43 In COMFORT-I, ruxolitinib was found to significantly decrease spleen size, ameliorate disease-related symptoms, and improve overall survival.42 In COMFORT II, 53% of ruxolitinib-treated patients had at least a 35% reduction in splenic volume and experienced a significant amelioration of constitutional symptoms. (improvement of 50% or more in the total symptom score at 24 weeks in 46% of patients).43 The COMFORT-II study also showed a trend toward a survival advantage after 5 years of follow up, but this was confounded by the trial's crossover design.43 Anemia and thrombocytopenia are the most common toxicities with ruxolitinib.
Other JAK inhibitors are being evaluated in clinical trials. The results of the PERSIST-1 phase 3 trial demonstrated that pacritinib was able to reduce spleen volumes and improve symptom burden in PMF patients with thrombocytopenia.44 Furthermore, one-fourth of patients who were red cell-transfusion dependent became transfusion independent on pacritinib. Another new JAK inhibitor, momelotinib, holds promise in improving anemia and transfusion requirements in patients with myelofibrosis. In the future, the clinical development of antifibrotic and antiangiogenesis therapies may play an important role in the therapeutic armamentarium.26
Patients asymptomatic at the time of diagnosis can have an indolent clinical course for several years. However, among the MPNs, PMF has the worst prognosis, with a median survival of 3.5 to 5.5 years.26 The most common causes of death were infection, cardiovascular disease, cerebrovascular disease, hemorrhage or thrombosis, and acute leukemia (blast-phase PMF). Patients with acute leukemia arising from PMF rarely achieve a remission from induction chemotherapy.
Essential thrombocythemia, previously called hemorrhagic thrombocythemia, is characterized by a sustained clonal proliferation of megakaryocytes in the bone marrow, with a peripheral blood platelet count greater than 600 x 109/L. This platelet count threshold was decreased to greater than 450 x 109/L in the 2008 WHO classification.2 Before making the diagnosis of ET, causes of reactive thrombocytosis must be excluded.
The prevalence of ET is estimated to be 38 to 57 per 100,000 population and is the lowest among patient with Philadelphia chromosome-negative MPNs.10,12 There may be a higher prevalence in younger women (approximately 2:1), and the median age at diagnosis is 60 years.
The proliferation of megakaryocytes is primarily caused by clonal stem cells, as confirmed by enzyme and genetic analysis.45 Megakaryocyte progenitor cells in ET are hypersensitive to the action of several cytokines, including IL-3 and IL-6, and possibly thrombopoietin.45 This leads to increased platelet production. There is controversy regarding spontaneous megakaryocyte formation in ET. Similar to primary myelofibrosis, mutations in JAK2 are found in 50% to 65% of ET cases.3 Patients lacking mutations in JAK2 may instead demonstrate activating mutations of the thrombopoietin receptor (MPL, 5% of patients) and calreticulin (CALR, 15 to 25% of patients).2,3
Increased platelet counts in ET are associated with increased thrombotic and hemorrhagic complications. Decreasing platelet counts in ET to below 600 x 109/L can decrease thrombotic complications.46 High platelet counts (>1,000 x 109/L) are associated with acquired von Willebrand disease resulting from the adsorption of von Willebrand multimers onto platelet membranes. A reduction in the platelet count is associated with correction of the defect and cessation of bleeding. Qualitative abnormalities in the platelets themselves are also likely to contribute to the increased risk of thrombotic and hemorrhagic complications in ET, because reactive thrombocytosis is not associated with an increased risk of thrombosis or bleeding, even with high platelet counts. Platelet aggregation studies in ET are often abnormal.
The clinical signs and symptoms are similar to those of PV. Many patients are asymptomatic at presentation, and their diagnosis is based on an elevated platelet count. However, patients can present with splenomegaly, hepatomegaly, or hemorrhagic or thrombotic episodes. From 13% to 37% of patients experience a hemorrhagic event, and 22% to 84% of patients experience a thromboembolic event.10 Hemorrhage is most common in the gastrointestinal tract. Constitutional symptoms, such as weight loss, fever, and pruritus, also can occur.
As with PV, thrombotic episodes can occur in the major vessels or microvasculature (see "Polycythemia Vera: Signs and Symptoms"). Thrombotic episodes occur more often in older patients and in patients with a history of thrombotic events. This increase in thrombotic risk with age has been attributed to the coexistence of vascular disease in older patients. Events tend to occur mainly in the microvasculature.
Women of childbearing age may have experienced a spontaneous abortion secondary to placental thrombosis, especially in the first trimester.
Essential thrombocythemia is described by persistent nonreactive thrombocytosis, a diagnosis of exclusion. The disease was first defined by the PV Study Group in 199746 and revised two decades later by the WHO. The WHO diagnosis requires patients to meet four criteria though additional changes to the diagnostic criteria have been proposed (Table 6).16
| Current (2008) WHO criteria | Proposed Criteria |
|---|---|
| Major criteria | |
|
|
| Minor criteria | |
|
|
| Requires all 4 major criteria | Requires all 4 major criteria OR first 3 major criteria and 1 minor criterion |
CML = chronic myelogenous leukemia; ET = essential thrombocythemia; MDS = myelodysplastic syndrome; PMF = primary myelofibrosis; PV = polycythemia vera; WHO = World Health Organization.
Adapted with permission from Tefferi A, Thiele J, Vannucchi AM, Barbui T. An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia 2014; 28:1407–1413.
The first step for any patient with thrombocytosis is to examine the peripheral blood film (Figure 5). Automated hematology analyzers can erroneously count platelet-sized particles that are red or white cell fragments as platelets (pseudothrombocytosis). Causes of reactive thrombocytosis include inflammatory states, infection, malignant disease, trauma, blood loss, and the postsplenectomy state. Other studies to obtain are iron studies to rule out iron deficiency; C-reactive protein, erythrocyte sedimentation rate, and fibrinogen to rule out inflammatory states; peripheral blood smear to rule out pseudothrombocytosis; and FISH or PCR testing for BCR/ABL to rule out CML.47
In a study of 280 patients with thrombocytosis, 82% of cases were reactive thrombocytosis.48 Some of these patients have platelet counts greater than 1,000 x 109/L, so the degree of thrombocytosis is not always helpful in making a diagnosis. Reactive thrombocytosis is associated with elevated levels of IL-6 or C-reactive protein in 81% of patients, which can help distinguish it from ET.48 Patients with uncomplicated thrombocytosis secondary to a myeloproliferative disorder typically have undetectable IL-6 levels.26
A bone marrow aspirate and biopsy with cytogenetics can be helpful in diagnosing ET and excluding other myeloid malignancies. Two-thirds of patients with ET have a bone marrow with marked megakaryocytic hyperplasia, morphologically bizarre megakaryocytes with nuclear pleomorphism, and clustering of megakaryocytes (Figure 6). Increased myeloid and erythroid precursors, abnormal cytogenetics, minimal reticulin fibrosis, and spontaneous megakaryocyte colony formation also may be present. These features are not typically present in the bone marrow of patients with reactive thrombocytosis.
Identifying a JAK2, MPL, or CALR mutation can be useful in excluding a reactive thrombocytosis. Additional clinical features that suggest ET rather than reactive thrombocytosis include a chronically elevated platelet count, splenomegaly, and history of thrombosis or hemorrhage. An elevated platelet count also may be seen in PV; the disorder would be considered PV if the other diagnostic criteria are met. Finding significant dyserythropoiesis or dysgranulopoiesis should prompt consideration of MDS rather than ET.
As with the other MPNs, treatment is based on risk stratification. High-risk patients include those older than 60 years of age or with a history of thrombosis. Low-risk patients include those younger than 60 years of age, no history of thrombosis or cardiovascular risk factors, and platelet counts ≤1,500 x 109/L. Intermediate-risk patients comprise the remainder of patients not falling into either of the above groups.49
Low-dose aspirin (81 mg) likely decreases the thrombotic risk in ET, although no randomized studies have been conducted as in PV. Patients with a platelet count greater than 1,500 x 109/L and acquired von Willebrand disease should avoid aspirin. Asymptomatic patients in the low-risk group can be followed with observation only. Patients in the high-risk group should be offered cytoreductive therapy. Controversy exists regarding the treatment of intermediate-risk patients.
Unlike the target hematocrit of under 45% for PV, a target platelet count does not exist for ET. A randomized study showed that hydroxyurea decreased the risk of thrombosis in high-risk patients who have ET from 24% to less than 4% (P=.003), compared with no treatment, when the platelet count is decreased to less than 600 x 109/L.46 Some have postulated that maintaining a platelet count less than 400 x 109/L may be associated with a further reduction in thrombosis, although this has not been confirmed in a randomized trial.45
In addition to hydroxyurea, anagrelide, which works by interfering with megakaryocyte maturation, can be used to control platelet counts in patients with ET. Anagrelide's main side effects include headaches, palpitations, and, rarely, a nonischemic cardiomyopathy. These effects are secondary to the drug's vasodilative inotropic actions; therefore, it should be used cautiously in patients with cardiac disease.45 Anagrelide can also decrease the hematocrit, but not the WBC count.
The United Kingdom Medical Research Council Primary Thrombocythemia 1 trial, a randomized controlled study, demonstrated that hydroxyurea should be considered first-line treatment for patients with high-risk ET.50 In this study, 809 patients were randomized to receive low-dose aspirin plus either anagrelide or hydroxyurea. Both anagrelide and hydroxyurea were able to control platelet counts, but the anagrelide recipients had significantly greater composite risk of arterial thrombosis, venous thrombosis, serious hemorrhage, or death from vascular causes. Anagrelide-treated patients also experienced a significantly greater increase in transformation to post-ET myelofibrosis (7% vs 2%) at 39 months of follow-up.
Another trial, the noninferiority ANAHYDRET study, concluded anagrelide was not inferior to hydroxyurea in preventing hemorrhagic or thrombotic complications in patients with ET.51 However, this study did not include aspirin as part of the treatment (only 28% were taking aspirin during this trial) making the results difficult to interpret.
Interferon-alfa is another option for patients, but its use in ET is often limited to cytoreduction in high-risk women of childbearing age.45 Cytoreduction is often used for ET during pregnancy because without treatment, up to 45% experience pregnancy loss. Pregnancy loss cannot be fully predicted by the course of disease, platelet count, or specific treatment. Anticoagulation therapy, with or without cytoreduction, can greatly reduce the risk of pregnancy loss. Agents such as 32P or busulfan are rarely used in ET, but they can be used in patients whose life expectancy is shorter than 10 years.45
Patients with life-threatening hemorrhagic or thrombotic events can be treated with thrombocytapheresis in combination with myelosuppressive therapy.52 Patients with a platelet count greater than 1,500 x 109/L and acquired von Willebrand disease should be treated with platelet-reduction therapy and avoid aspirin. These patients also might require treatment with factor VIII concentrates that contain von Willebrand factor and desmopressin in the setting of a bleeding episode. Finally, patients undergoing surgery are at an increased risk of thrombotic and bleeding episodes. If possible, their platelet counts should be normalized before surgery.
Because life expectancy for patients with ET approaches that of the normal population, the prognosis largely depends on the patient's age and history of thrombosis. The International Prognostic Score for Essential Thrombocythemia analyzed results from a cohort of 891 patients to develop a point system corresponding to level of risk (Table 7).53
| Parameter | Points |
|---|---|
| Age ≥60 years | 2 |
| WBC count ≥11 x 109/L | 1 |
| History of thrombosis | 1 |
| Total points | Risk group | Median survival, years |
|---|---|---|
| 4 | High | 8.8 |
| 3 | High | 16.5 |
| 2 | Intermediate | 19.9 |
| 1 | Intermediate | 24.5 |
| 0 | Low | Not Reached |
Data from Passamonti F, et al. Blood 2012; 120:1197–201.
Scores based on age, white blood cell count, and history of thrombosis corresponds to high, intermediate, and low risk groups.53 Few patients with ET transform to acute myeloid leukemia (blast phase), and approximately 5% develop myelofibrosis.53
Michael Keng, MD, Karl Theil, MD, and Anjali Advani, MD, contributed to previous versions of this topic review.
Aaron Gerds, MD, disclosed consulting / advisory fees from Incyle Corp.