
Published: August 2017
Pulmonary hypertension (PH) is elevated blood pressure in the pulmonary artery (PA) averaging 25 mm Hg or above at rest.1 Elevated PA pressure (PAP) can be caused by abnormalities in the precapillary pulmonary arterioles, called pulmonary arterial hypertension (PAH), or by abnormalities that increase left atrial pressure resulting in back pressure on the pulmonary circulation, called pulmonary venous hypertension (PVH). Although the etiology and pathophysiology of PAH and PVH are distinct, they are similar in that both cause elevated PA pressure that inevitably leads to right ventricular (RV) dilation and remodeling, followed by RV failure and death if RV compensation can no longer sustain normal cardiac function.
The clinical features of PAH were first described by Paul Wood in 1952, though the first pathology-based classification system for PH occurred in 1973 in the wake of a European epidemic of PAH associated with use of anorexigens.2,3 The World Health Organization (WHO) established 2 types of PH: primary PH (PPH), characterized by the absence of identifiable causes or risk factors; and secondary PH, characterized by the presence of identifiable causes or risk factors. The diagnosis of PPH is one of exclusion and is known today as idiopathic PAH (IPAH). The PH classification system has been revised many times, most recently in 2013 at the 5th World Symposium held in Nice, France, which recognized a classification system for PH introduced in 2003.4,5 The system classifies PH into 5 groups based on pathophysiology and clinical features. These are identified in Table 1.5
| 1. Pulmonary arterial hypertension (PAH) |
|---|
1.2. Heritable PAH
1.2.2. ACVRL1 (ALK1), ENG, SMAD9, CAV1, KCNK3 1.2.3. Unknown 1.4. Associated with
1.4.2. HIV infection 1.4.3. Portal hypertension 1.4.4. Congenital heart diseases 1.4.5. Schistosomiasis 1' Pulmonary veno-occlusive disease (PVOD) and/or pulmonary capillary hemangiomatosis (PCH) |
| 2. Pulmonary hypertension owing to left heart diseases |
2.2. Diastolic dysfunction 2.3. Valvular disease 2.4. Congenital / acquired left heart inflow/ outflow tract obstruction and congenital cardiomyopathies. |
| 3. Pulmonary hypertension owing to lung diseases and/or hypoxia |
3.2. Interstitial lung disease 3.3. Other pulmonary diseases with mixed restrictive and obstructive pattern 3.4. Sleep-disordered breathing 3.5. Alveolar hypoventilation disorders 3.6. Chronic exposure to high altitude 3.7. Developmental lung disease |
| 4. Chronic thromboembolic pulmonary hypertension (CTEPH) |
| 5. Miscellaneous |
5.2. Systemic disorders (sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis) 5.3. Metabolic disorders (glycogen storage disease, Gaucher disease, thyroid disorders) 5.4. Others (tumoral obstruction, fibrosing mediastinitis, chronic renal failure, segmental pulmonary hypertension) |
BMPR = bone morphogenic protein receptor type II; CAV1 = caveolin-1; ENG = endoglin; HIV = human immunodeficiency virus.
Adapted from Simonneau G, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62:D34-D41. ©2013 with permission from the American College of Cardiology Foundation. http://www.sciencedirect.com/science/journal/07351097.
The main focus of this review is WHO group 1 PAH. Within the discussion, the terms anorexigen-associated PAH (APAH), familial PAH (FPAH), and idiopathic PAH (IPAH) will be used to describe distinct entities within the WHO group 1 classification. The term PAH will be used to describe WHO group 1 PAH as a category, unless specified otherwise.
The diagnosis of PAH is increasing worldwide, but it remains a rare disease. The prevalence ranges from 15 to 52 cases per million population and the annual incidence ranges from 2.4 to 7.6 cases per million population.6 PAH predominately effects middle-aged females. Initial reports from the National Institutes of Health in the 1980s reported the mean age at diagnosis of PAH was 36 years and the female-to-male ratio was 1.7 to 1.7 Interestingly, the age at diagnosis and gender distribution of PAH may be evolving as more recent studies report a mean age at diagnosis of 50 years and higher female-to-male ratios.8,9 The reason for these shifts remains unclear.
Despite greater awareness of PAH, the current median interval from the onset of symptoms to diagnosis remains unacceptably high at 13.6 months, unchanged from the 1980s.9 That said, the general consensus is that improvements in survival have occurred due to evolving definitions of PAH, earlier identification of PAH in patients, and new therapies for PAH.10 Before the use of pulmonary vasodilators, patients diagnosed with PAH had a median survival of 2 to 3 years.11 The Registry to Evaluate Early And Long-term PAH Disease Management (REVEAL registry) data suggests that patients with newly diagnosed PAH are living longer, reporting 3-year survival estimates of 69% and 5-year survival estimates of 61.2%.12
Our understanding of the relationship between genetic mutations and PAH has increased in the past 20 years. In the United States, REVEAL registry data shows 3.1% (64 of 2,039) of patients with previously diagnosed PAH have a first-degree relative with PAH.12 The most common genetic abnormalities occur in the BMPR2 gene and there are over 300 independent BMPR2 mutations. BMPR2 mutations have been found in 75% of patients with a known family history of PAH and in 26% of patients without a known family history of PAH.13-15 The BMPR2 gene encodes a protein involved in the control of vascular cell proliferation and appears to be inherited as an autosomal dominant trait with a variable genetic penetrance, dependent on the gender of the patient. The Vanderbilt Pulmonary Hypertension Registry reported 42% penetrance in females and 14% penetrance in males.16 Contrary to prior assertions, anticipation, whereby a genetic disorder passed to the next generation manifests at an earlier age and in a more severe form, is not supported by the current FPAH data.
Other genetic causes of PAH include mutations in ACVRL1, SMAD9, ENG, CAV1, and KCNK3 genes.13 The prevalence of these mutations is lower than BMPR2 and genetic screening is generally reserved for patients with hereditary hemorrhagic telangiectasia (ACVRL1 and ENG), who present at a very young age with IPAH or who have a family history of PAH but no evidence of BMPR2 mutation.10
More recently, whole genome sequencing has demonstrated biallelic mutations in the EIF2AK4 gene in all familial cases of pulmonary veno-occlusive disease (PVOD) and pulmonary capillary hemangiomatosis (PCH). This mutation is also present in 25% of all sporadic PVOD and PCH cases.17 Prior to this discovery, PVOD and PCH could only be confirmed via histologic analysis, a dangerous precedent as these patients have a high risk of pulmonary hemorrhage following lung biopsy. Recent guidelines recommend that patients suspected of PVOD and PCH be tested for a biallelic mutation of EIF2AK4 and that a positive finding is sufficient to confirm the diagnosis and forgo lung biopsy.10
PAH is a proliferative vasculopathy affecting distal pulmonary arteries and arterioles less than 50 micrometers in diameter. The histologic profile generally includes medial hypertrophy, eccentric or concentric fibrotic intimal lesions with or without the presence of organized thrombi, and destructive plexiform lesions.18
Over the past decade, considerable research has been done to better understand the pathogenesis of PAH. Key factors in PAH development are in part related to an imbalance between locally produced vasodilation mediators, such as nitric oxide and prostacyclin (PGI2), and vasoconstriction mediators, such as endothelin and thromboxane. The physiological role of these mediators has largely been supported by improved patient outcomes with use of therapies targeting these pathways.
Nitric oxide is an endothelium-derived relaxing factor that plays a pivotal role in the pathobiology of IPAH. A potent pulmonary vasodilator, nitric oxide produced locally in the lungs has profound effects on smooth muscle relaxation and proliferation, maintaining the normal pulmonary vascular tone. The close proximity of the airways to the blood vessels in the lungs allows nitric oxide that is produced in high levels in the upper and lower airways by nitric oxide synthase 2 to affect the pulmonary vascular tone in concert with the low nitric oxide levels that are produced by nitric oxide synthase 3 in the vascular endothelium. Patients with IPAH have low levels of nitric oxide in their exhaled breath and the severity of the disease inversely correlates with nitric oxide reaction products in bronchoalveolar lavage fluid.19-21
Inhaled nitric oxide would be a fantastic therapeutic modality were it not for the cost and technical difficulties to administer it. As such, use of nitric oxide has been limited to the vasoreactivity testing during pulmonary artery catheterization.
Endothelin-1 is a potent vasoconstrictive peptide secreted by the vascular endothelium that contributes to the proliferation of surrounding vascular smooth muscle cells. The pulmonary circulation plays an important role in the production and clearance of endothelin-1 and this physiologic balance is reflected in the circulating levels of endothelin-1. Patients with PAH, IPAH in particular, have an increased expression of endothelin-1 in pulmonary vascular endothelial cells and a concomitant increase in serum endothelin-1 levels.22,23
The endothelium also produces prostacyclin, which is metabolized from arachidonic acid by the cyclooxygenase pathway. Prostacyclin causes vasodilation throughout the circulation and is an inhibitor of platelet aggregation by its action on platelet adenylate cyclase. The final enzyme in the production of prostacyclin is prostacyclin synthase. The remodeled pulmonary vasculature in lung tissue obtained from patients with severe IPAH expresses low levels of prostacyclin synthase when compared with normal lung tissue. In addition, prostacyclin metabolites are diminished in the urine of patients with PH, further emphasizing the role of prostacyclin in this disease.24,25
In addition to vasoconstriction, pulmonary vascular remodeling plays a major role in the increased pulmonary vascular resistance (PVR) seen in IPAH. The same paracrine mediators that cause abnormal vasoconstriction are responsible for abnormalities of mitogenic (ie, endothelin-1) and antimitogenic (ie, prostacyclin and nitric oxide) origin that lead to proliferative dysregulation. These abnormalities have also been noted in neoplastic processes.26 Several findings support this theory:
Like many neoplastic processes, endothelial cells undergo metabolic changes consistent with the Warburg phenomenon.27 The Warburg phenomenon occurs when cells preferentially synthesize pyruvate to lactate via anaerobic metabolism pathways despite adequate oxygen being present in the cytoplasm. This phenomenon also results in decreased mitochondrial activity and a decline in nitric oxide production. In malignancy, the Warburg phenomenon allows for cellular proliferation to continue even with an insufficient vascular supply of oxygenation. While access to adequate vascular supply is not necessarily an issue in PAH, the metabolic changes occurring are consistent with a pseudoneoplastic process.
IPAH is associated with irreversible plexogenic lesions formed by abnormally proliferating monoclonal endothelial cells. This suggests that random somatic mutations are responsible for initiating the development of PAH. Because of metabolic reprogramming of these cells, certain conditions including hypoxia would selectively benefit the growth of these cells.
The BMPR2 gene is involved in regulation of multiple functions including cellular proliferation, differentiation, apoptosis, and endothelial–mesenchymal transition.28 Loss of function mutations, such as that seen in BMPR2, permits excess unchecked endothelial cell growth through dysregulation of apoptotic pathways within endothelial cells of the pulmonary vasculature. CAV1, another rare mutation in PAH, causes dysfunction of the membrane protein involved in cell proliferation and nitric oxide signaling.13
PAH has variable penetrance in familial cases. Exposure to external factors such as viral infections (eg, human immunodeficiency virus [HIV]), inflammation (eg, autoimmune disorders), hypoxic insults (eg, obstructive sleep apnea), or toxins (eg, cocaine, amphetamines, anorexigenic medications) may provide the second hit required for disease presentation similar to cancer pathogenesis. Targeting genetic and epigenomic factors such as microRNA has been postulated as a possible future treatment.29
Another important pathologic feature of PAH is in situ thrombosis within diseased pulmonary arterioles. Whether this is a consequence of or a cause of vasculopathy remains debatable. The increased propensity for thrombosis arises at the microvascular level where dysfunctional endothelium loses the anticoagulant properties that usually prevent intravascular clotting of blood. Coagulopathy mediators that are usually inhibited in normal individuals become activated. In addition, prostacyclin and nitric oxide, both inhibitors of platelet aggregation, are decreased at the level of the injured endothelial cell. Circulating platelets in patients with PAH seem to be in a continuous state of activation, further contributing to coagulopathy. They also promote endothelial remodeling and smooth muscle proliferation by releasing thromboxane A2 and serotonin.
Finally, abnormalities in the extracellular matrix also contribute to the medial hypertrophy commonly seen in pulmonary arterioles of patients with PAH. Through a dynamic process of matrix protein degradation and synthesis, triggered by the high flow and pressure in the pulmonary vasculature, the extracellular matrix is remodeled contributing to the obliterative changes seen in the pulmonary arteries.30
The pathobiology of PAH is more complex than previously believed. A better understanding of the pathogenesis of PAH will lead to new therapeutic interventions to improve outcomes for patients with this disease.
The symptoms of PAH are nonspecific, which leads to delays in evaluation and diagnosis. The symptoms are invariably related to right ventricular dysfunction: patients typically present with exertional symptoms such as dyspnea, chest pressure, and lightheadedness. As the disease progresses, symptoms occur at rest too as the ability of the right ventricle fails to compensate against increased PVR.
In addition to dyspnea, symptoms of PAH also include fatigue, weakness, angina, lightheadedness or syncope, lower extremity edema, and abdominal distention.10,31 The clinical suspicion of PH should arise in any case of dyspnea without overt signs of specific heart or lung disease. Patients with underlying lung or heart disease, as well as those with conditions frequently linked to PAH (connective tissue diseases, cirrhosis of the liver, HIV infection, and congenital heart disease), who develop dyspnea or other symptoms associated with right ventricle dysfunction should be screened for PAH.10,31
The physical signs of PH also reflect changes in right ventricle function and structure. Cardiac findings include left parasternal lift, loud pulmonary component of the second heart sound, pansystolic murmur of tricuspid regurgitation, diastolic murmur of pulmonary insufficiency, and right ventricular third heart sound. Extracardiac signs include jugular vein distention, hepatomegaly, peripheral edema, and ascites. The lung examination is usually normal.
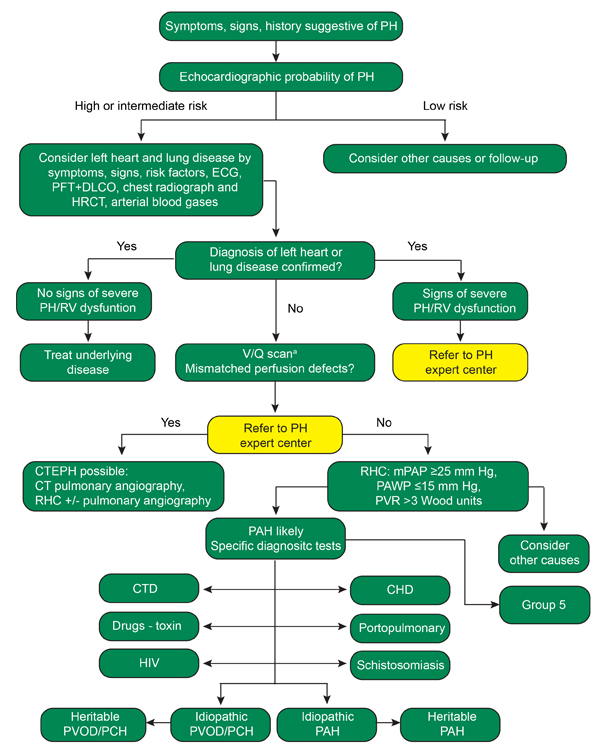
Once suspected, the diagnosis of PAH requires a series of investigations to confirm the diagnosis and identify an optimal therapeutic approach (Figure 1).10 Patients suspected of having PAH should undergo an echocardiogram and the findings should be used by the clinician to clarify the patient’s risk of having WHO Group 1 PAH or another form of PH.
Pulmonary artery systolic pressure can be estimated using echocardiography with the simplified Bernoulli's equation:
Right ventricular systolic pressure = [4 x (tricuspid regurgitant velocity)2 + right atrial pressure]
Generally, pulmonary artery systolic pressure greater than 40 mm Hg is considered abnormal and should prompt further investigation. However, among individual patients, right ventricular systolic pressure does not correlate well with systolic PAP measured by right heart catheter.32
Therefore, other echocardiographic parameters should be employed to improve the accuracy of risk stratification (Table 2).10
Low risk |
Intermediate risk |
High risk |
|||
|---|---|---|---|---|---|
| Peak tricuspid regurgitant velocity, m/s | ≤2.8 |
<2.8 |
2.9 to 3.4 |
2.9 to 3.4 |
>3.4 |
| Right ventricular systolic pressure, mm Hg | 35 |
<35 |
36 to 50 |
36 to 50 |
>50 |
| Other echocardiogram variables suggestive of pulmonary hypertension | No |
Yes |
No or few |
Yes |
|
Adapted with permission of the European Society of Cardiology & European Respiratory Society ©. European Respiratory Journal Oct 2015, 46 (4) 903-975; DOI: 10.1183/13993003.01032-2015. All rights reserved. © 2015 European Society of Cardiology & European Respiratory Society.
Other echocardiographic parameters that are suggestive of RV dysfunction and PH include
If echocardiographic evaluation reveals an intermediate or high risk of PH, further investigation of a PH etiology should be pursued. The most common cause of PH is left ventricle dysfunction and aortic or mitral valve disease. If left ventricle dysfunction or significant aortic/mitral valve disease is noted, further workup for the cause of PH is not explicitly warranted and efforts should focus on the management of the underlying left ventricle or valvular disorder.
Pulmonary function testing and computed tomography imaging may reveal underlying parenchymal lung disease as a cause of PH in patients without left ventricle or valve dysfunction. It is also essential to rule out the presence of chronic thromboembolic disease in patients with suspected PAH. Although image quality of computed tomography pulmonary angiography (CTPA) has improved considerably, ventilation-perfusion (VQ) scanning is recommended over CTPA because VQ scan sensitivity is near 100%.34 Unfortunately, this is not true of CTPA given the differences in radiographic appearance between chronic and acute thromboembolism and the poor inter-reader reliability among radiologists for detecting chronic pulmonary embolism. In patients with a positive VQ scan or patients with suspected chronic thromboembolic pulmonary hypertension (CTEPH), referral to an expert center for evaluation of surgical pulmonary thromboendarterectomy is warranted as this is currently the only curable form of PH.35 Patients who with perfusion abnormalities on VQ scan should be referred to a PH center of excellence for further evaluation of CTEPH surgical management.
If left ventricle dysfunction and parenchymal lung disease are not suspected causes of PH and VQ scan is negative, right heart catheterization (RHC) should be pursued. RHC is required for the diagnosis of WHO group 1 PAH and it is used for management and prognostication.
Pulmonary hypertension is present when the mean PAP is ≥25 mm Hg. In the absence of CTEPH or significant lung or left heart disease, WHO Group 1 PAH is defined as mean PAP ≥25 mm Hg in association with pulmonary artery occlusion pressure (PAOP) ≤15 mm Hg and a PVR > 3 Wood units. When PH is diagnosed, but the PAOP is > 15 mm Hg, one should suspect a postcapillary etiology for PH such as left ventricular dysfunction or mitral/aortic valve disease. Differentiation of PAH from postcapillary PH has important prognostic and treatment implications.
Of all the RHC determinants, PAOP is subject to the greatest error in measurement and interpretation, so careful attention is needed when obtaining this measurement or reviewing a RHC report. Five criteria are used to evaluate whether a PAOP measurement is valid:
If PAOP is not reliable, then left ventricular end-diastolic pressure should be directly measured.
A small subset of patients with PAH demonstrate remarkable reactivity to pulmonary vasodilators during RHC. This population has an excellent prognosis and unlike most patients with PAH, responds well to calcium channel blocker therapy. In all patients with suspected IPAH, FPAH or APAH, vasoreactivity testing should be performed during RHC with inhaled nitric oxide. Alternatively, intravenous epoprostenol, inhaled iloprost, or adenosine are also suitable for determining pulmonary vasoreactivity. If vasoreactivity testing elicits a drop in mean PAP of ≥10 mm Hg to an absolute level ≤40 mm Hg, with a preserved or increased cardiac output, a positive test is observed.5 This occurs in approximately 10% of patients with IPAH. In these patients, it is generally considered safe to initiate high-dose calcium channel blocker therapy. However, approximately half of these patients will become nonreactive over time. Therefore, frequent reassessment with RHC every 6 to 12 months should be pursued to ensure long-term calcium channel blocker response. In patients who become nonreactive or who exhibit symptoms consistent with World Health Organization functional class (WHO-FC) III or IV, a switch to conventional PAH treatment is warranted.10
Once PAH is confirmed by RHC, further evaluation of secondary causes should be pursued. This may include serologic testing to exclude HIV, thyroid disorders, or connective tissue disease, especially scleroderma. In cases where FPAH is suspected, genetic testing is warranted. Ultrasound of the liver to exclude cirrhosis and redoubled efforts at eliciting a thorough past medical history from the patient regarding prior toxin/anorexigen exposure is also important. Overnight oximetry or polysomnography is useful in detecting obstructive sleep apnea contributing to PH since obstructive sleep apnea is highly prevalent in patients with PAH.37,38
The treatment goal for patients with PAH is to improve right ventricular function to minimize mortality risk (Table 3).10 This is achieved both pharmacologically and with lifestyle modifications. As defined in the 2015 ERS guidelines for diagnosis and management of PH, treatment of patients with PAH involves 3 major aspects of intervention:
| Determinants of prognosisa (estimated 1-year mortality) |
Low risk
(< 5%)
|
Intermediate risk (5–10%) |
High risk
(>10%)
|
|---|---|---|---|
| Clinical signs of right heart failure | Absent | Absent | Present |
| Progression of symptoms | No | Slow | Rapid |
| Syncope | No | Occasional syncopeb | Repeated syncopec |
| WHO functional class | I, II | III | IV |
| 6MWD | > 440 m | 165–440 m | < 165 m |
| Cardiopulmonary exercise testing | Peak VO2 >15 mL/min/kg (>65% pred.) VE/VCO2 slope <36 |
Peak VO2 11–15 mL/min/kg (35–65% pred.) VE/VCO2 slope 36–44.9 | Peak VO2 <11 mL/min/kg (<35% pred.) VE/VCO2 slope ≥45 |
| NT-proBNP plasma levels | BNP <50 ng/L NT-proBNP <300 ng/L | BNP 50–300 ng/L NT-proBNP 300–1400 ng/L |
BNP >300 ng/L NT-proBNP >1400 ng/L |
| Imaging (echocardiography, CMR imaging) |
RA area <18 cm2 No pericardial effusion |
RA area 18–26 cm2 No or minimal, pericardial effusion |
RA area >26 cm2 pericardial effusion |
| Hemodynamics | RAP <8 mm Hg CI ≥2.5 L/min/m2 SvO2 >65% |
RAP 8–14 mm Hg CI 2.0–2.4 L/min/m2 SvO2 60-65% |
RAP >14 mm Hg CI <2.0 L/min/m2 SvO2 <60% |
6MWD: 6-minute walking distance; BNP: brain natriuretic peptide; CI: cardiac index; CMR: cardiac magnetic resonance; NT-proBNP: N-terminal pro-brain natriuretic peptide; pred.: predicted; RA: right atrium; RAP: right atrial pressure; SvO2: mixed venous oxygen saturation; VE/VCO2: ventilatory equivalents for carbon dioxide; VO2: oxygen consumption; WHO: World Health Organization.
aMost of the proposed variables and cut-off values are based on expert opinion. They may provide prognostic information and may be used to guide therapeutic decisions, but application to individual patients must be done carefully. One must also note that most of these variables have been validated mostly for IPAH and the cut-off levels used above may not necessarily apply to other forms of PAH. Furthermore, the use of approved therapies and their influence on the variables should be considered in the evaluation of the risk.
bOccasional syncope during brisk or heavy exercise, or occasional orthostatic syncope in an otherwise stable patient.
cRepeated episodes of syncope, even with little or regular physical activity.
Reproduced with permission of the European Society of Cardiology & European Respiratory Society ©. European Respiratory Journal Oct 2015, 46 (4) 903-975; DOI: 10.1183/13993003.01032-2015. All rights reserved. © 2015 European Society of Cardiology & European Respiratory Society.
Treatment strategies and determination of an individual’s prognosis depend largely on the severity of PAH and the clinical assessment forms the cornerstone of this evaluation. History and physical examination at every visit should address medication compliance and symptoms consistent with right ventricular dysfunction. All patients should undergo an assessment of functional capacity at the time of PAH diagnosis and at each follow-up visit. This includes assessment of WHO-FC, a simple yet highly prognostic parameter,39,41 and either a 6-minute walk test (6MWT) or cardiopulmonary exercise test (CPET).
The 6MWT is economical and easy to perform. Absolute values achieved during a 6MWT rather than between-test changes provide better prognostic information.42 Many thresholds have been proposed, but no specific threshold is optimal for every patient. For example, patients with scleroderma may exhibit lower baseline walk distances compared with patients with IPAH given the high prevalence of myopathy associated with scleroderma. In addition to other factors (Table 3), current guidelines state a 6MWT distance >440 meters is optimal and is one of several factors associated with a low 1-year mortality.10,43
CPET is more costly and cumbersome but can provide significant insight into a patient’s cardiopulmonary physiology. When CPET is utilized, management decisions are most commonly based on peak oxygen consumption (VO2) values. Patients with peak VO2 < 11 mL/min/kg (<35% predicted) and ventilator equivalents for carbon dioxide produced (VE/VCO2) slope ≥45 is suggestive of a high mortality risk.10
Other prognostic factors are based on evaluation of right ventricular function. Tests to evaluate right ventricular function include biomarkers associated with myocardial stress or cardiovascular imaging such as cardiac magnetic resonance imaging (cMRI) and echocardiography. Two specific biomarkers, B-type natriuretic peptide (BNP) and N-terminal pro b-type natriuretic peptide (NT-proBNP), are excreted by the right and left ventricles. Increased levels are associated with worsening myocardial stress and dysfunction. While increased levels of BNP/ NT-proBNP cannot differentiate between right ventricle and left ventricle dysfunction, these biomarkers are helpful in the longitudinal monitoring of PH if utilized appropriately. In PAH, elevated levels of BNP/ NT-proBNP are associated with increased mortality. Decreased levels following initiation of therapy are correlated with a favorable prognosis.44
Echocardiography allows direct visualization of right ventricular structures and estimation of PAP. However, estimated right ventricular systolic pressure provides little prognostic information and should not be used alone to inform therapeutic decisions. Instead, a detailed evaluation of right ventricular anatomy and function should be obtained. Changes consistent with right ventricular dysfunction, including increasing right atrial or right ventricular size, increased left ventricular eccentricity index, a decrease in tricuspid annular plane systolic excursion, or a new pericardial effusion should prompt further evaluation for worsening disease.45,46 Exercise echocardiogram can provide further useful data regarding right ventricular function. The ability of the right ventricle to generate an exercise induced increase in systolic PAP of greater than 30 mm Hg is shown to be an independent marker of survival in severe PH.47
In recent years, cMRI has gained favor because it can provide a superior assessment of right ventricular morphology and function compared with echocardiography. In addition, the prognostic utility of cMRI seems promising.48 However, poor tolerance by patients, cost, and the lack of accurate PAP estimates with cMRI has hampered its widespread use.
The optimal interval to obtain longitudinal testing depends on the patient’s clinical stability and physician preference. While guidelines exist, these are largely based on expert opinion. At our center, most patients follow up with their PH physician every 3 to 6 months and have a 6MWT and BNP or NT-proBNP test at every visit. Echocardiography is generally obtained every 6 to 12 months and right heart catheterization is usually performed when a patient’s clinical status changes or with initiation of a new medication. Table 4 provides recommendations regarding follow up testing for patients with PAH.10
| At baseline | Every 3-6 monthsa | Every 6-12 monthsa
|
Every 3-6 months after change in therapya |
In case of clinical worsening
|
|
|---|---|---|---|---|---|
| Medical assessment and determination of functional class | + |
+ |
+ |
+ |
+ |
| Electrocardiogram | + |
+ |
+ |
+ |
+ |
| 6-minute walk test /Borg dyspnea scale | + |
+ |
+ |
+ |
+ |
| Cardiopulmonary exercise testing | + |
+ |
+e |
||
| Echocardiography | + |
+ |
+ |
+ |
|
| Basic labb | + |
+ |
+ |
+ |
+ |
| Extended labc | + |
+ |
+ |
||
| Blood gas analysisd | + |
+ |
+ |
+ |
|
| Right heart catheterization | + |
+f |
+e |
+e |
aIntervals to be adjusted according to patient needs.
bBasic lab includes blood count, international normalized ratio (in patients receiving vitamin K antagonists), serum creatinine, sodium, potassium, aspartate aminotransferase / alanine aminotransferase (in patients receiving endothelin receptor antagonists), bilirubin and brain natriuretic peptide / N-terminal pro-brain natriuretic peptide.
cExtended lab includes thyroid stimulating hormone, troponin, uric acid, iron status (iron, ferritin, soluble transferrin receptor) and other variables according to individual patient needs.
dFrom arterial or arterialized capillary blood; may be replaced by peripheral oxygen saturation in stable patients or if blood gas analysis is not available.
eShould be considered.
fSome centers perform right heart catheterization at regular intervals during follow-up.
Reproduced with permission of the European Society of Cardiology & European Respiratory Society ©. European Respiratory Journal Oct 2015, 46 (4) 903-975; DOI: 10.1183/13993003.01032-2015. All rights reserved. © 2015 European Society of Cardiology & European Respiratory Society.
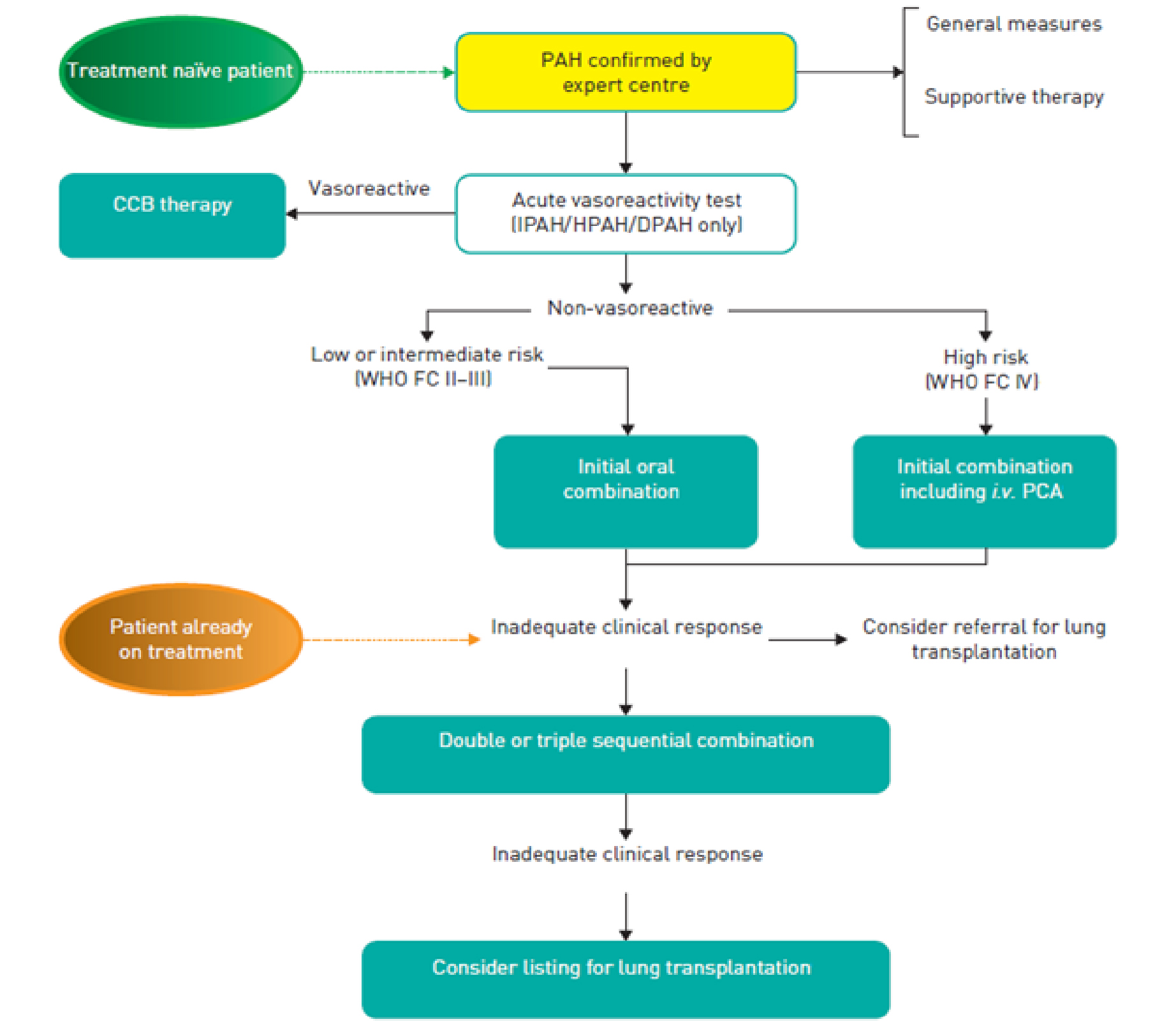
Choice of specific agents should be guided by the severity of PAH, patient symptoms, baseline hemodynamics, and clinical mortality risk (Figure 2).10 In general, high-dose calcium channel blockers (CCB) are contraindicated in the treatment of PAH and should only be used in the rare patient with IPAH, FPAH or anorexogen associated PAH who meets criteria for vasoreactivity during RHC. The vast majority of patients with WHO group 1 PAH (regardless of vasoreactive status) should be started on one or more pulmonary vasodilators chosen from the 3 principal vasodilator pathways currently available. In patients with WHO-FC I, II, or III, there is no consensus on which of pathway is optimal as a first line therapy, but there is general agreement that a combination regimen including at least two different classes of therapy should be prescribed. Combination therapy has been shown to improve outcomes when compared to monotherapy in multiple trials.49,50 In patients at high risk of PAH, intravenous, subcutaneous, or inhaled prostacycline analogs should be considered as part of the initial combination therapy. Assessment of treatment response should be based on objective criteria as discussed in the treatment goal section.
Patients with IPAH who exhibit acute vasoreactivity during RHC have improved survival with long-term use of CCB.51 Thus, these agents should be reserved for patients who have significant and definite responses to a short-acting vasodilator during RHC. Unfortunately, a very small proportion (approximately 5%) of IPAH patients qualify for and benefit from long-term therapy with oral CCB. In patients who are vasoresponsive, the calcium channel blocker doses required for efficacy are much higher than traditional doses; nifedipine (120 mg to 240 mg), diltiazem (240 mg to 720 mg) and amlodipine (20 mg). Because these medications can elicit significant hypotensive effects, they should be up titrated carefully. Of note, vasodilator responsiveness is not uniformly associated with a good long term response to calcium channel blocker in all patients with PAH. Patients with HIV, connective tissues diseases, portopulmonary hypertension, and pulmonary veno-occlusive disease or pulmonary capillary hemangiomatosis who demonstrate vasoresponsiveness during RHC do not exhibit favorable prognosis similar to patients with IPAH, FPAH, and anorexogen-associated PAH. Thus vasoreactivity testing is discouraged and CCB contraindicated in these populations.
Epoprostenol (Flolan, Veletri) is a synthetic analogue of prostacyclin delivered by continuous intravenous infusion that improves exercise capacity, hemodynamic variables, and survival in patients with IPAH and is the treatment of choice for severely ill patients.52 In the past, epoprostenol therapy was complicated by the instability of the drug at room temperature and the need for continuous intravenous infusion because of the drug's short half-life. More recently, inhaled, subcutaneous, and oral prostacyclin formulations have been developed as well as longer acting, room-temperature stable formulations. However, these newer formulations have not demonstrated mortality benefits similar to those of earlier epoprostenol trials. This may be due to inferiority of newer prostacyclin formulations or the use of background therapy in patients with PAH in more recent clinical trials. Thus, adding prostacyclin to background pulmonary vasodilators, while beneficial, may not offer the profound effect seen in original trials and has not demonstrated improved patient outcomes.53,54
Common side effects of prostacyclin analogues are headache, flushing, jaw pain, diarrhea, nausea, skin rash, and musculoskeletal pain. In addition, there is no optimal delivery option for prostacyclins. Intravenous formulations can deliver a sustained therapeutic dose but have risks of catheter associated infection and thrombus and the physical and social limitations inherent with continuous infusion therapy. Subcutaneous formulations often cause severe pain and skin ulceration at the infusion site, which may be intolerable and lead to discontinuation. Oral treprostinil (Orenitram) is conveniently administered, but has only demonstrated efficacy as a monotherapy. Two inhaled prostacyclin analogues, iloprost (Ventavis) and treprostinil (Tyvaso), are currently approved by the U.S. Food and Drug Administration (FDA) for patients with IPAH. Due to the relatively short duration of action, these medications must be used several times daily to maintain a therapeutic effect (treprostinil 4 times/day and iloprost 6 to 9 times/day).
Selexipag (Uptravi) is a prostacyclin receptor agonist that is chemically dissimilar from other prostacyclin analogs in that it targets the prostacyclin receptor, which is often downregulated in PAH. It is available as an oral preparation and has been found to be efficacious as monotherapy or in combination with endothelin receptor antagonists (ERAs) and/or phosphodiesterase type 5 (PDE 5) inhibitors.55 Selexipag carries a similar side effect profile to conventional prostacyclin therapy and patients should be advised of them before starting selexipag.
Three ERAs are currently approved by the FDA. Bosentan (Tracleer), an orally administered dual type A and type B ERA, is approved on the basis that it improved 6MWT distance, functional class, and time to clinical worsening in patients with PAH. Patients using bosentan are at risk of asymptomatic increase in liver enzyme levels and thus should have liver function monitored at least monthly.
Ambrisentan (Letairis) is a selective type A ERA shown to improve functional class, walking distance, and reduce time to clinical worsening in patients with PAH.56 Ambrisentan does not require monthly monitoring of liver function tests. Macitentan (Opsumit) is another dual type A and type B ERA that significantly decreased a composite endpoint of morbidity and mortality. Although the significance of the endpoint was largely driven by reductions in morbidity (not mortality), macitentan is generally well tolerated, and like ambrisentan, is not associated with liver function abnormalities.
Other potential side effects of ERAs include edema, teratogenicity, testicular atrophy, and male infertility. ERAs are contraindicated in pregnancy due to their teratogenic effects and the FDA requires that all patients who take ERAs should be counseled to use 2 forms of birth control and are required to undergo monthly pregnancy testing.
There are currently 2 approaches to therapeutic approaches that target the nitric oxide pathway. The phosphodiesterase type 5 (PDE 5) inhibitors, sildenafil (Revatio) and tadalafil (Adcirca) are currently approved by the FDA for use in patients with PAH.57 By inhibiting PDE 5, these medications stabilize cyclic guanosine monophosphate (cGMP, the second messenger of nitric oxide), allowing a more sustained effect of endogenous nitric oxide, an indirect but effective and practical way of using the nitric oxide-cGMP pathway.
While conventional PDE 5 inhibitors augment the nitric oxide–cGMP pathway by slowing cGMP degradation, soluble guanylate cyclase (SGC) stimulators enhance cGMP production. Riociguat (Adempas) is a SGC stimulator introduced in the U.S. in 2013 that improves exercise capacity, hemodynamics, WHO-FC, and time to clinical worsening. More recently, in an open-label multicenter, single-arm study, patients currently on a PDE 5 inhibitor were switched to riociguat under the hypothesis that SGC stimulation would provide additional pulmonary vasodilator response and thus improved downstream outcomes.58 An interim analysis suggested lower pulmonary vascular resistance, increased cardiac output, and improved 6MWT distances overall. This data has resulted in the advent of a randomized international, multicenter, double arm; open label trial to assess the efficacy of riociguat compared with conventional PDE 5 inhibitor therapy in patients with PAH.59 Riociguat is also effective in the medical treatment of recurrent or inoperable CTEPH.60,61 Because PDE 5 inhibitors and riociguat target different areas of the same pathway, combination therapy is contraindicated. Major side effects include hypotension, edema, and syncope. Although PDE 5 inhibitors are not contraindicated in patients of childbearing age, riociguat is contraindicated in pregnancy due to potential teratogenic effects. As with ERAs, all patients who take riociguat should be counseled to use 2 forms of birth control and are required to undergo monthly pregnancy testing.
Balloon atrial septostomy to treat PAH is increasingly uncommon with the increased availability of other therapeutic options. However, it might be beneficial in the setting of severe disease with recurrent syncope or right heart failure (or both) despite maximal medical therapy. The procedure can also be used as a bridge to lung transplantation. The rationale for its use is that the controlled creation of an atrial septal defect would allow right-to-left shunting, leading to increased systemic output and systemic oxygen transport despite the accompanying fall in systemic arterial oxygen saturation. The shunt at the atrial level also allows for decompression of the right atrium and right ventricle, alleviating some of the effects of right heart failure. Balloon atrial septostomy is a high-risk procedure and should be performed only in experienced centers.10
Lung transplantation has been used in treatment for PH since the 1980s. It is indicated in patients with advanced PAH that is refractory to available medical therapy. Single and bilateral lung transplantations have been performed for IPAH, but most transplant centers currently perform bilateral lung transplantation to minimize postoperative complications. Survival rates after lung and heart-lung transplantation are approximately 52% to 75% at 5 years and 45% to 66% at 10 years.62,63,64
Pulmonary thromboendarterectomy (PTE) provides a potential surgical cure for patients with CTEPH. The procedure involves dissecting well-organized thromboembolic material from the intimal layer of the pulmonary arterial bed. Patients with suspected CTEPH should be referred to centers with surgical experience in treating CTEPH for consideration of PTE. In patients with operable CTEPH, PTE is the treatment of choice because it improves hemodynamics, functional status, and survival. In patients who are deemed inoperable, a second opinion should be obtained. In patients who have inoperable CTEPH or who experience residual PH following PTE, pulmonary vasodilator therapy is indicated. In patients with inoperable or residual CTEPH, riociguat has been shown to improve 6MWT distances and decrease PVR and has been approved specifically for this population.
Historically, physical activity has been discouraged in patients with PAH under the premise that increased PA pressures during exercise would further impair RV function and lead to poor outcomes. However, several studies have challenged this notion and a meta-analysis by Pandey et al suggests that exercise training in PAH is associated with improved outcomes and carries little risk of adverse effect.65 In light of this, our institution promotes supervised exercise in patients with PAH through pulmonary rehabilitation training. Patients should exercise to a degree where they are not experiencing significant symptoms of RV dysfunction such as chest pain or lightheadedness. Resistance training, in which patients often perform breath holding or Valsalva maneuvers is discouraged. Severely deconditioned patients should be started on pharmacological treatment prior to initiating exercise training.10,66
Pregnancy should be avoided in patients with PAH, as pregnancy and delivery is associated with a high risk of maternal and fetal mortality. Although successful pregnancies have been reported and recent reports suggest better outcomes than previously suggested, maternal mortality remains near 20%.67 Two forms of birth control are recommended for all patients of child-bearing potential. At present, there is no agreement on the most appropriate birth control method in patients with PAH. Since there is an increased risk of thrombosis with estrogen-based contraception, progesterone containing preparations may be considered, but use of endothelin receptor antagonists such as bosentan can reduce the efficacy of these agents. Insertion of an intrauterine device is another option; however, the rare side effect of a vasovagal response during insertion could result in cardiovascular collapse in a patient with PAH.68 Barrier methods and sterilization should also be considered along with their benefits and side effects, including the risk of operative complications and ectopic pregnancies. Safety of hormonal therapy in postmenopausal women is still unclear. It may be considered in extreme cases if combined with oral anticoagulant therapy. Patients taking riociguat or endothelin receptor antagonists must undergo monthly pregnancy testing as these medications carry significant teratogenic risk.
When efforts at pregnancy prevention fail, patients should be advised regarding the high risk of maternal and fetal death. In cases of continued pregnancy, PAH medications should be limited to PDE 5 inhibitors such as sildenafil or tadalafil, or prostacyclin medications (treprostenil or epoprostenol) during pregnancy or lactation.69
Elective surgery involves an increased risk of cardiopulmonary collapse in patients with PAH. The risk is proportional to the severity of the disease and is generally related to the ability of the RV to tolerate perioperative fluid shifts and changes in sympathetic tone and pleural pressure following endotracheal intubation associated with general anesthesia. Local and regional anesthesia is advised when feasible. If general anesthesia is required, surgery should be performed at a PH referral center with experienced anesthesia and PH teams that can anticipate and respond to complications.70,71 Depending on the type of surgery, oral PAH medication may need to be transitioned to intravenous or inhaled forms during the perioperative period. Anticoagulant treatment should be interrupted for as short a period as possible, but generally does not require bridging, with the exception of patients with CTEPH where bridging with heparin is recommended to minimize risk of further thrombus formation.
Patients with PAH are susceptible to developing lower respiratory tract infections. Primary prevention with pneumococcal vaccination and yearly influenza shots is recommended and should be offered to all patients.9,10
PAH is a severe, debilitating disease with significant psychological, emotional, social, and financial impacts on patients and their families.72 Availability of appropriate referral services and support groups to help these patients is essential. Local and national PAH support groups are common and may be found on the internet or through a patients PH provider. The Pulmonary Hypertension Association (PHA) is perhaps the largest national support group and offers local, regional, and national support services in addition to conferences, videos, and online and printed patient education. All patients should be evaluated to ensure appropriate social support at home. In addition, early advanced care planning is an essential element of high-quality PH treatment. In cases where symptoms of RV dysfunction are progressive despite optimal therapy, referral to palliative care services should be considered as an adjunct to ongoing PH treatment efforts.10
Since hypoxia can worsen vasoconstriction in patients with PAH, it is best to avoid or prepare for situations where hypoxia may occur. This includes travel to high altitude locations and air travel.73 Supplemental oxygen should be considered for patients with baseline resting oxygen saturation <95% on room air, who are WHO-FC III or IV or who have a room air partial pressure of dissolved oxygen less than 60 mm Hg. Because commercial airlines only allow patients to carry portable oxygen concentrators onboard, those who are currently on oxygen or who meet the above criteria should undergo flight simulation testing prior to anticipated air travel or to altitudes above 1,500 meters.74
The pathophysiology of IPAH includes formation of microvascular thrombotic lesions. This insight together with early, small studies indicating benefits of anticoagulation in patients with PAH resulted in broad agreement that anticoagulation was beneficial.75 However, recognition of several methodologic flaws in early studies and evidence from larger, observational, registry-based studies has called the therapeutic benefits into question, leaving patients and providers in a state of continued equipoise.76,77 As such, use of anticoagulation in PH centers is variable and consensus remains based largely on expert opinion. Nonetheless, there is agreement that any potential benefit of anticoagulation is largely confined to patients with IPAH, HPAH, and PAH related to anorexigen. In patients with systemic sclerosis, at least 2 large observational trials have suggested increased harm with use of anticoagulants.77 In addition, among providers who use anticoagulants in patients, the target international normalized ratio varies among U.S providers (1.5 to 2.5) compared with European providers (2 to 3). There has been growing interest in the use of new oral anticoagulants in PAH; however, these agents have not been adequately studied in this population and are not currently recommended.
Hypoxemia is associated with pulmonary vasoconstriction, however evidence of the long-term benefit of supplemental oxygen is lacking. Guidelines for oxygen use in patients with PAH are largely extrapolated from those used in chronic obstructive pulmonary disease (COPD). While the standard practice in patients with PAH or COPD who demonstrate resting or exertional hypoxemia has largely been to offer supplemental oxygen, the recent Long Term Oxygen Treatment Trial has challenged this practice. In this trial, patients with stable COPD and moderate resting hypoxemia (oxygen satuation at rest 80% to 93%) or exertional hypoxemia (oxygen saturation nadir 80% to 90% during 6MWD) did not show improved mortality or time to hospitalization when supplemental oxygen was provided.78 At present we still offer oxygen to all patients with a resting or exertional oxygen saturation less than 89% or nocturnal hypoxemia as assessed by polysomnography or home sleep study under the premise that this prevents additional pulmonary arterial vasoconstriction. However, further studies specific to PAH are needed to clarify guidelines regarding the benefit of supplemental oxygen in this population.
Diuretics are widely prescribed in patients with PAH and are indicated to maintain optimal RV performance. In addition, diuretics can improve peripheral fluid retention in the abdomen, pleural space, and lower extremities. While diuresis is indicated to prevent volume overload in most patients with PAH, diuretics should be used with caution. Since patients with PAH and RV dysfunction maintain a state of preload dependence, over diuresis can cause diminished RV stroke volume and output, resulting in downstream systemic hypotension, arrhythmias, or shock. In addition, diuretics are associated with electrolyte changes including hyponatremia and hypokalemia and alkalosis. Electrolytes should be routinely monitored in patients with PAH who are using diuretics.
Digoxin has not been well studied in patients with WHO group 1 PAH, but is often used in patients with RV failure in order to improve RV function and to control supraventricular tachycardia. In patients using digoxin, care should be taken to avoid digitalis toxicity if used. The long-term effects of digoxin in these patients remains unknown and is recommended on the basis of expert opinion for patients with refractory RV failure and for rate control in PAH patients with atrial flutter or fibrillation.10
PAH is a well-recognized complication of chronic liver disease and cirrhosis. Portal hypertension rather than the liver disease itself seems to be the main determining risk factor for developing PH. The mechanism remains unclear but the presence of portosystemic shunt might allow vasoconstrictive and vasoproliferative substances, normally cleared by the liver, to reach the pulmonary circulation. Portopulmonary hypertension can be seen in up to 10% of patients evaluated for liver transplantation.79 Thus, echocardiographic screening to detect PH in patients with liver disease is appropriate in patients with symptoms and is recommended in candidates for liver transplantation.
Pulmonary artery catheterization should be performed in all patients with increased right ventricular pressure on echocardiography. Hemodynamically, patients with portopulmonary hypertension might have a significantly higher cardiac output and significantly lower systemic vascular resistance than patients with other forms of PAH. The treatment of portopulmonary hypertension can be challenging and is not well standardized. The general approach is similar to that in IPAH, except that anticoagulation is not recommended because of the risk of bleeding, and bosentan is avoided because of the potential for hepatoxicity.79 Other ERAs are less hepatotoxic, but have not been systematically evaluated in this population, thus caution is advised. Beta-blockers, which are frequently used in portal hypertension, should be avoided due to concerns of worsening hemodynamics and exercise capacity in these patients.
Mild cases of portopulmonary hypertension with mean PAP less than 35 mm Hg generally do well with liver transplantation with reversal of PH posttransplant. However, severe and uncontrolled PH is associated with high posttransplant mortality. There is anecdotal evidence that suggests treatment and appropriate response to PH therapy can allow these patients to undergo liver transplantation with improved clinical outcomes.
Pulmonary hypertension in the setting of chronic hypoxia due to underlying lung disease represents a challenging area for evaluation and management. Although chronic hypoxia is a recognized cause of PH, it rarely leads to severe PH. Nonetheless, patients with advanced COPD and interstitial lung disease who develop PH have worse outcomes.80 Thus, patients with chronic hypoxia who have a marked elevation in PAP (>35 mm Hg) should be evaluated for other causes of the PH.
To date, data supporting the benefit of pulmonary vasodilators in patients with PH due to chronic hypoxia is lacking. However, some experts suggest that PH therapy may be beneficial in patients with PA pressures elevated beyond what would be expected for their lung disease alone, but this too is not based on robust studies. Nonetheless, current recommendations based on expert opinion suggest that when mean PAP is greater than 35 mm Hg, it should be considered out of proportion to underlying lung disease.10 In all cases of PH due to chronic hypoxia, the first-line therapy is supplemental oxygen. When mild PH is associated with obstructive sleep apnea, the first line therapy should be treatment of the sleep apnea followed by evaluation of PH. If PH persists despite adequate therapy for sleep apnea, consider treating the PH as a separate disease.
{kind=link}
{kind=link}